






支鏈氨基酸轉氨酶2在癌細胞中調節鐵死亡
2020年10月江蘇大學附屬醫院Haitao Zhu教授于Cell Death & Differentiation上發表文章Branched-chain amino acid aminotransferase 2 regulates ferroptotic cell death in cancer cells其研究發現確認了BCAT2在鐵死亡中的新作用,提示了一種克服索拉非尼耐藥的潛在治療策略。

鐵死亡是一種由細胞氧化還原穩態喪失引起的細胞凋亡,導致脂質過氧化失控,最終導致細胞死亡。胱氨酸-谷氨酸抗氧化劑(系統Xc)或谷胱甘肽過氧化物酶4 (GPX4)的藥理失活可誘導鐵死亡增多,提示谷胱甘肽依賴的抗氧化防御在防止鐵死亡細胞死亡中起關鍵作用。鐵死亡作用與缺血引起的器官損傷、與退行性疾病相關的病理細胞死亡以及不同類型的癌癥有關。多種腫瘤細胞易發生鐵死亡增多,包括淋巴瘤、宮頸癌、頭頸癌、胰腺癌、腎細胞癌和肝細胞癌。各種研究證實了鐵亡誘導劑的關鍵作用,包括小分子鐵亡誘導劑如伊拉斯汀,以及許多藥物(如索拉非尼、青蒿素及其衍生物)在殺傷腫瘤細胞和抑制腫瘤方面的作用增長。這些鐵死亡誘導劑還可與化療藥物協同治療腫瘤。有趣的是,有些類型的癌癥對鐵死亡誘導劑比其他類型的更敏感。反向過硫通路已被確定為鐵死亡作用的負調節因子,卵巢癌細胞中缺乏該通路與伊拉斯汀誘導的鐵死亡敏感性增高有關。HSF1-HSPB1通路也在人宮頸癌、前列腺癌和骨肉瘤中負調控星抑素誘導的鐵死亡作用。MUC1-C/xCT通路是伊拉斯汀誘導的三陰性乳腺癌細胞鐵胞作用的另一個負調控因子。
越來越多的證據表明,細胞代謝在鐵死亡中起著至關重要的作用[12,13]。轉錄因子NRF2協調抗氧化防御系統調節鐵死亡作用。p62- Keap1-NRF2是[14]肝癌細胞上鐵死亡的中樞抑制通路。基因或藥物抑制NRF2顯著增強鐵死亡 伊拉斯汀引起的肝癌易感性和索拉非尼,而NRF2表達式led的激活細胞抵抗鐵死亡,建議中央部分含氧降低分子的作用,特別是活性氧(ROS)在鐵死亡。細胞內的鐵代謝對鐵依賴氧化酶或芬頓化學作用也是必不可少的。最近的一項研究表明,鐵死亡的自噬降解通過自噬貨物受體核受體共激活因子4 (NCOA4)調節鐵死亡的作用。不足為奇的是,氨基酸代謝也參與了鐵死亡。高濃度的胞外谷氨酸,伊拉斯汀,或其他系統Xc抑制劑阻斷胞內胱氨酸/半胱氨酸攝取,以誘導鐵死亡。沉默cysteinyltRNA合成酶(CARS)可上調經硫途徑,從而導致對伊拉斯汀誘導的鐵死亡的抗性。谷氨酰胺通過其特定的代謝酶谷氨酰胺酶(GLS1和GLS2)介導鐵死亡作用,但谷氨酰胺分解過程的機制復雜。然而,在肝癌細胞中控制鐵死亡敏感性的代謝途徑尚不清楚。在本研究中,作者鑒定了支鏈氨基酸氨基轉移酶2 (BCAT2),一種氨基轉移酶催化硫氨基酸代謝,作為鐵死亡的特異性抑制劑。作者發現BCAT2參與了Xc系統抑制物誘導的肝癌細胞鐵死亡作用。此外,BCAT2還參與了磺胺嘧啶與索拉非尼協同誘導鐵死亡的機制。因此,作者的研究結果表明BCAT2是鐵死亡的抑制因子,并參與了肝癌鐵死亡的核心代謝信號通路。
在這項研究中,作者證實了伊拉斯汀、索拉非尼或磺胺嘧啶激活了鐵死亡,增加了細胞不穩定鐵的水平。細胞內高水平的不穩定鐵導致細胞內ROS的快速積累,這是鐵中毒的必要條件。有趣的是,作者發現該噬鐵途徑也激活AMPK磷酸化,從而抑制SREBP1的核易位,并抑制其直接靶基因BCAT2的轉錄(圖5)。作者進一步發現,BCAT2通過調節細胞內谷氨酸水平抑制了細胞內的鐵死亡。重要的是,在體外和一些動物模型中,如皮下胰腺癌模型、原位肝癌模型和PDX肝癌模型中,硫柳氮嗪與索拉非尼的聯合在抑制BCAT2表達和促進鐵毒性癌細胞死亡方面具有協同作用。重要的是,在這些臨床前癌癥模型中,BCAT2也顯示出作為評估藥物反應的敏感生物標志物的潛力。BCAT2控制鐵死亡的發現與氨基酸在[21]鐵死亡中起關鍵作用的概念一致。BCAT是關鍵的代謝蛋白,催化BCAA的可逆轉氨基反應生成各自的a-酮酸(BCKAs)和谷氨酸,負責生成30%的從頭腦谷氨酸。谷氨酸的代謝與鐵死亡的調控密切相關。值得注意的是,Xc系統的功能是由谷氨酸水平調節的,因為在Xc系統中谷氨酸以1:1的比例交換胱氨酸。因此,細胞外高濃度的谷氨酸阻斷系統Xc活性,抑制胱氨酸攝取,并驅動鐵死亡。相比之下,作者體外實驗中細胞內高水平的谷氨酸來源于BCAT2驅動的谷氨酸的重新合成,從而可能增強系統Xc活性,促進胱氨酸攝取,抑制鐵亡。BCAT2細胞內谷氨酸代謝的這種保護作用與細胞外腦谷氨酸水平降低對Xc敲除小鼠神經毒性損傷的保護作用是一致的。此外,作者的實驗證實了BCAT2的表達水平受到AMPK-SREBP1信號通路的調控,這也得到了之前在胰腺癌[18]中BCAT2基因組缺失導致側死的發現的支持。作為代謝應激的關鍵傳感器,AMPK在鐵死亡作用中的作用與環境有關。AMPK通過介導BECN1磷酸化和BECN1- slc7a11復合物的形成,從而促進癌細胞的鐵死亡,這與作者發現的鐵死亡抑制劑通過激活AMPK而下調BCAT2表達的研究結果一致。有趣的是,作者注意到最近的報道,能量應力介導的AMPK激活抑制了鐵死亡作用。AMPK在鐵死亡中作用的差異可能與被測細胞系的基礎AMPK活性和環境背景有關。因此,AMPK在癌癥鐵死亡中的潛在作用值得深入研究,也為腫瘤患者提供了一種重要的治療策略。
BCAA是合成谷氨酸和谷氨酰胺的氮供體,但谷氨酰胺在鐵死亡中的作用是復雜的。谷氨酰胺是通過其特殊的代謝-谷氨酰胺分解來降解的。當glutaminolysis抑制或谷氨酰胺耗盡,胱氨酸饑餓和阻斷胱氨酸進口均不能誘導ROS積累、脂質過氧化和鐵死亡增多,說明谷氨酰胺溶解是鐵死亡增多的燃料。根據這一觀察,除谷氨酰胺外,谷氨酰胺分解的另一產物a-酮戊二酸(a-KG)可替代谷氨酰胺誘導鐵死亡。作者推測BCAT催化BCAAs-BCKAs穿梭合成谷氨酸,導致細胞內a- kg水平降低,這可能是引起鐵死亡的另一個原因。敲除BCAT2對SLC7A11的表達無影響。因此,BCAT2與系統Xc之間的確切關系需要在今后的工作中進行更深入的研究。肝癌是全球癌癥死亡的第三大原因,而標準的化療對大多數肝癌患者并沒有效果,醫生一直在尋找靶向療法。索拉非尼是唯一的多激酶抑制劑作為一線治療被證明可以延長不可切除的HCC[26]的總生存率。然而,來自亞太地區服用索拉非尼的患者的總生存率僅為6.5個月,反應率低為2%。
最近,最近的一項三期臨床研究中,其總體生存優勢并沒有超過索拉非尼。在這項研究中,作者發現磺胺嘧啶單獨或與索拉非尼聯合,在二鐵誘導療法中發揮作用。這些發現是相應的,因為磺胺嘧啶是一種抗炎藥物,已經廣泛用于炎癥性腸病的慢性、長期治療,保證了其在成人和兒童中的安全性。基于作者的研究結果,磺胺嘧啶可能是晚期肝癌以及其他無法切除的癌癥的一種潛在的新治療選擇。由于治療中表達的變化,BCAT2可能是最敏感的靶點之一,其表達可作為預測索拉非尼和磺胺嘧啶聯合治療反應的標志物。然而,這一假設應該在患者資料中進行評估。綜上所述,作者的數據表明,抑制細胞內谷氨酸的合成可以作為在癌癥背景下誘導鐵死亡依賴性的一個好策略。作者的研究結果支持了這一觀點,即硫磺胺嘧啶與索拉非尼協同下調BCAT2,從而下調細胞內的谷氨酸。作者的工作還表明,細胞殺傷機制涉及調控從頭合成谷氨酸是肝癌細胞的關鍵過程。作者認為BCAT2的蛋白或mRNA水平可用于預測癌細胞對未來鐵運誘導療法的反應性。作者還提出,高特異性的BCAT2抑制劑可以為一部分有意義的癌癥患者提供有效的治療。
技術路線:
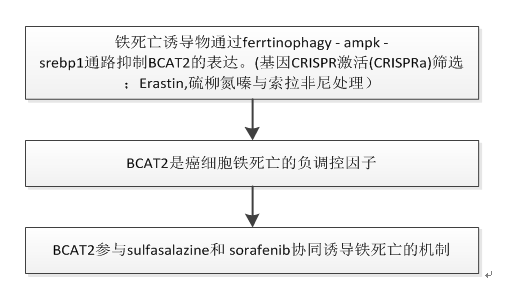
一、用kinome CRISPRa篩選鑒定鐵死亡的新作用以及Ferroptosis誘導物通過ferritinophagy-AMPK-SREBP1通路抑制BCAT2的表達

A. 為了系統闡明ferroptosis下游保守負調控因子,我們進行了大規模的基因CRISPR激活(CRISPRa)篩選。靶向2320激酶、磷酸酶和藥物靶點(https://www.addgene.org/ pooled-library/weissman-human CRISPRa -v2-subpools/)的pooled人CRISPRa sgRNA慢病毒庫和Cas9-VPR酶通過慢病毒轉導引入HepG2細胞,然后用erastin處理或對照DMSO處理.富集sgRNAs的基因靶點是在HepG2細胞中賦予erastin介導的ferroptosis抗性的潛在基因。在我們的篩選方法中,許多報道的ferroptosis基因被鑒定和驗證。其中分支的BCAT2是ferroptosis潛在負調控因子的首選基因.
B. 在ferroptosis誘導劑處理后,BCAT2的蛋白和mrna表達水平有所下降,但BCAT1 (BCAT2的同源物)沒有下降,
在甲磺酸二鐵胺(DFO,一種ferroptosis抑制劑)的存在下被逆轉。
C. 推測ferroptosis誘變劑通過AMPK-SREBP1信號通路下調BCAT2。通過western blotting定量分析,erastin、sorafenib或s ulfasalazine誘導了蘇氨酸殘基172 (T172)上AMPK磷酸化,并降低了SREBP1的表達(圖1C、S4A和S4B)。
D. ChIP實驗還顯示,erastin、sorafenib或sulfasalazine存在時,SREBP1與BCAT2的結合顯著減少.
E. AMPK激活劑AICAR下調Aspc-1和HepG2癌細胞中BCAT2的表達方式與erastin、sorafenib或sulfasalazine類似,AMPK抑制劑化合物C可逆轉這一過程,進一步證實了ferroptosis誘變劑通過激活AMPK下調BCAT2的表達.
二、BCAT2是癌細胞鐵死亡的抑制因子
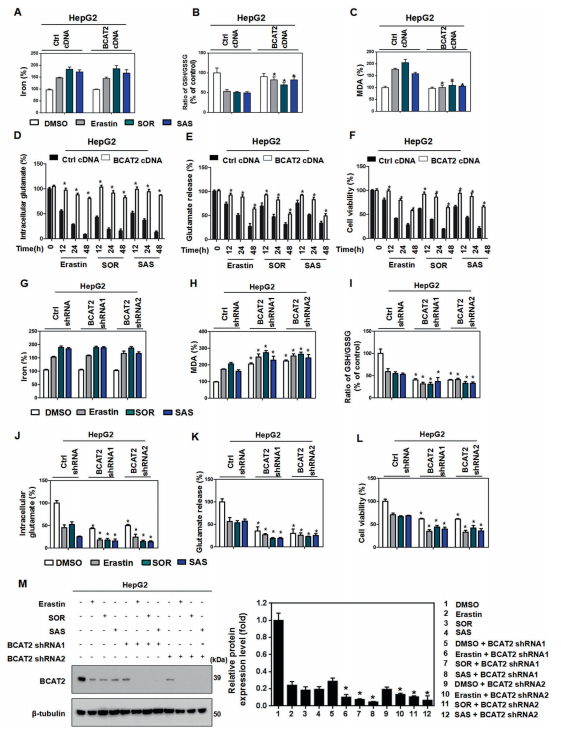
A.考慮到鐵在鐵死亡中的關鍵作用,我們首先研究了BCAT2表達與鐵積累的相關性。
與預期的一樣,erastin、sorafenib或sulfasalazine處理在對照組和BCAT2轉染的細胞中都誘導了游離鐵的積累.
B. erastin, sorafenib, and sulfasalazine 處理后,Aspc-1和HepG2細胞的GSH水平受到抑制,BCAT2的異位表達恢復了GSH水平.
C.與親代細胞相比,erastin, sorafenib, and sulfasalazine處理后,脂質過氧化的終產物丙二醛(MDA)水平在Aspc- 1和HepG2細胞中升高,而過表達bcat2的細胞中降低(圖2C和S8D)。
D-F.與這些結果一致,在erastin, sorafenib, and sulfasalazine存在下,BCAT2過表達增加了細胞內谷氨酸(圖2D和S8E)和谷氨酸釋放(圖2E和S8F),并以時間依賴的方式減少了 system Xc抑制劑誘導的細胞死亡(圖2F和S8G).
G-L.在erastin, sorafenib, and sulfasalazine存在的情況下,敲除BCAT2-顯著增加了Aspc-1和HepG2細胞中MDA的生成(圖2H和S11D)和GSH的消耗(圖2I和S11E),但對游離細胞鐵的積累沒有影響(圖2G和S11C)。此外,BCAT2在erastin, sorafenib, and sulfasalazine存在下,下調細胞內谷氨酸水平(圖2J和S11F),谷氨酸釋放(圖2K和S11G)以及細胞活力(圖2L和S11H)。用RSL3和BSO處理親代和BCAT2敲除Aspc-1和HepG2細胞,顯示出相似的結果(圖S12)。因此,BCAT2沉默細胞的集落形成能力受到抑制(圖S11I)。敲除BCAT2并不影響Aspc-1和HepG2細胞中SLC7A11和GPX4蛋白的表達水平(圖S13A)。這些結果表明,BCAT2敲低可部分誘導鐵死亡。
三、BCAT2參與sulfasalazine和 sorafenib協同誘導鐵死亡的機制
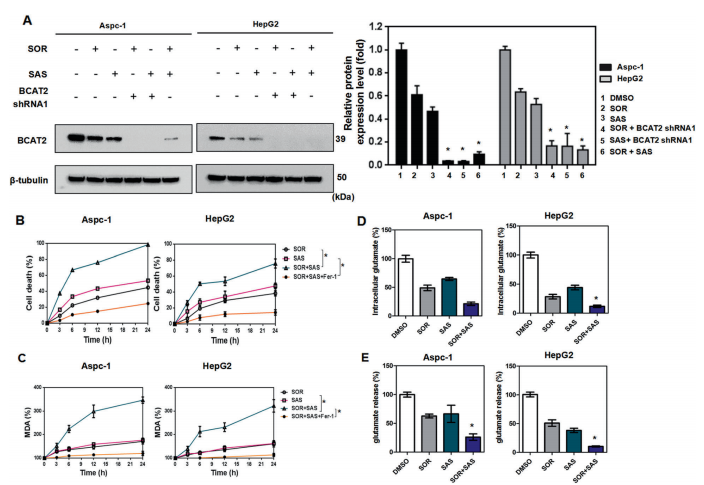
A-E.sorafenib and sulfasalazine聯合應用可顯著抑制Aspc-1和HepG2細胞中BCAT2的表達(圖3A),與sorafenib and sulfasalazine單獨聯合BCAT2 shRNA的模式相似(圖2M和S11J)。sorafenib and sulfasalazine在增加細胞死亡(圖3B)和MDA生成方面也表現出協同作用,在ferrostatin-1 存在時可以恢復這一作用(圖3C),抑制細胞內谷氨酸水平(圖3D),谷氨酸釋放(圖3E)。
總之,sorafenib and sulfasalazine通過部分調節BCAT2表達對ferroptosis的影響。
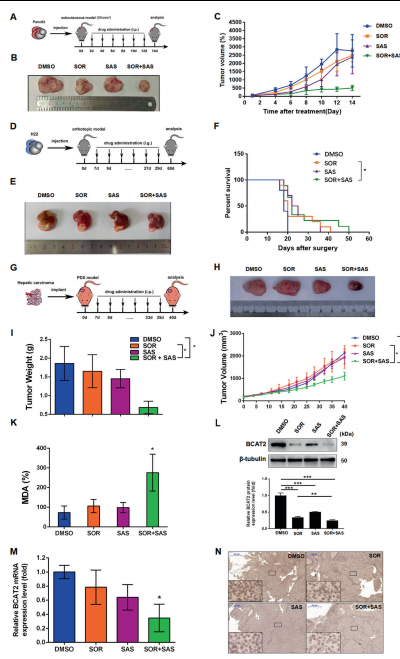
在C57BL/6小鼠中,在C57BL/6小鼠中,sorafenib and sulfasalazine分別減少了9.63%和13.5%的Panc02皮下腫瘤大小,聯合治療在第14天進一步減少了81.39%的腫瘤大小(圖4A-C)。
由于原位異種移植模型在復制腫瘤微環境和預測藥物療效方面被認為優于皮下腫瘤模型,
在C57BL/6小鼠H22細胞的原位HCC模型中,我們想驗證 sulfasalazine誘導鐵死亡是否也能增強索拉非尼的抗癌活性(圖4D)。
sorafenib and sulfasalazine顯著減小了原位異種移植瘤的腫瘤大小(圖4E),延長了動物存活時間(圖4F)。
此外,sorafenib and sulfasalazine聯合治療顯著降低了蛋白和mrna表達水平中的BCAT2(圖s)。S14A和S14B),
原位HCC組織中GSH水平降低(圖S14C), MDA水平升高(圖S14D)。在C57BL/6小鼠H22細胞的原位HCC模型中,我們想驗證磺胺吡啶誘導二茂鐵tosis是否也能增強索拉非尼的抗癌活性(圖4D)。的確,索拉非尼聯合柳氮磺胺吡啶顯著減小了原位異種移植瘤的腫瘤大小(圖4E),延長了動物存活時間(圖4F)。
此外,索拉非尼和柳氮磺胺嘧啶聯合治療顯著降低了蛋白和mrna表達水平中的BCAT2(圖s)。S14A和S14B),原位HCC組織中GSH水平降低(圖S14C), MDA水平升高(圖S14D)。
為了揭示更多的臨床相關性,我們轉向patient-derived異種移植(pdx)模型,已應用于臨床前藥物檢測在許多類型的癌癥由于其生物穩定,準確反映病人腫瘤組織病理學、基因表達、基因突變,和治療反應(4G).