






人臍帶血來源的MSCs外泌體通過抑制鐵死亡緩解急性心肌梗死小鼠心肌損傷
急性心肌梗死(Acute myocardial infarction, AMI)常因氧和血供突然減少而引起心肌損傷和心力衰竭,是發病和死亡的主要原因之一。鐵死亡是一種氧化的、依賴鐵的細胞死亡形式,不同于壞死、凋亡和自噬。

細胞內谷胱甘肽(GSH)依賴的抗氧化防御系統失活,導致毒性脂質活性氧(ROS)的積累,從而導致鐵細胞膜下垂。據報道,鐵tosis可發生在缺血再灌注(I/R)損傷、癌癥等。鐵死亡是心肌細胞死亡的一種重要形式。ferroptosis在急性心肌梗死中起重要作用。人臍帶間充質干細胞(hub -MSCs)因其免疫原性低、增殖因子高、轉染效率高,具有廣闊的應用前景。有報道稱,HUCBMSCs來源的外泌體緩解了AMI損傷。那么,間充質干細胞外泌體是否對AMI中ferroptosis有調節作用呢?二價金屬轉運體DMT1 (Slc11a2)作為復雜生理過程的關鍵組成部分,可以調節體內鐵的水平。在哺乳動物中,dmt1是一種鐵(2+)的轉運體,對適當維持鐵的穩態至關重要。有鑒于此,河南省人民醫院,阜外中華心血管病醫院麻醉科的Yufang Song、河南省人民醫院阜外中國中部心血管醫院心血管外科Zhenwei Ge等研究人員,探討DMT1在AMI體外和體內模型中的表達及作用。
據報道,人臍血來源的MSCs外泌體對急性心肌梗死(AMI)和心肌細胞缺氧損傷小鼠模型有心肌保護作用,但具體機制有待進一步研究。
本文旨在研究hucb - msc -外泌體抑制鐵死亡減輕心肌損傷的作用。RT-PCR和western blotting結果顯示,與假手術組和常氧組比較,二價金屬轉運體1 (DMT1)表達顯著升高,Prussian藍染色、鐵離子(Fe2+)、MDA和GSH水平檢測結果顯示,缺氧損傷后心肌梗死和心肌細胞出現鐵死亡。過表達DMT1促進H/ r誘導的心肌細胞鐵死亡,而敲除DMT1則顯著抑制了心肌細胞鐵死亡。hub - msc來源的外泌體抑制了鐵死亡和減少心肌損傷,miR-23a- 3p敲除后在外泌體中消失。雙熒光素酶報告基因實驗證實DMT1是miR-23a-3p的靶基因。綜上所述,hucb - mscs -外泌體可能通過miR-23a-3p抑制DMT1的表達,從而抑制鐵死亡,減輕心肌損傷。
技術路線:

結果:
一、在AMI小鼠中上調DMT1的表達并增加鐵死亡

B.建立小鼠AMI模型,分別于術后1 h、4 h、12 h、24 h、48 h采集左心室心肌組織。RT-PCR和western blot結果顯示,AMI小鼠24 h和48 h時DMT1表達明顯高于假手術組,而GPX4蛋白表達降低.表明AMI模型建立后24 h DMT1明顯上調。
C.使用Prussian藍染色和鐵檢測試劑盒,AMI小鼠在12 h、24 h和48 h鐵沉積和亞鐵(Fe2+)水平增加(圖1c、d)。
D.AMI小鼠24 h和48 h MDA水平升高,GSH水平和GPX4活性均下降。提示AMI模型LAD后24 h可出現心肌組織鐵死亡。

A-B.用H/R處理新生小鼠心肌細胞,并轉染攜帶DMT1表達序列/DMT1干擾序列或陰性對照的慢病毒。與對照組相比,H/R組DMT1 mRNA和蛋白表達水平均上調(圖2a, b)。
C. 在H/R處理下,細胞活力測定和形態學的變化顯示,過表達DMT1進一步降低了細胞活力,而敲除DMT1則明顯提高了細胞活力(圖2c)。
D. 正常心肌細胞偽足相互接觸呈放射狀排列,H/R處理使細胞突觸變薄、收縮、胞漿萎縮。在H/R處理下,DMT1過表達進一步引起細胞凋亡,核染色質凝集碎裂,細胞收縮,而DMT1敲除明顯改善細胞質收縮(圖2d)。
G,H. 流式細胞儀分析數據顯示,H/R誘導細胞凋亡,過表達DMT1進一步加重細胞凋亡,而敲除DMT1則顯著抑制細胞凋亡.
F,J-K . H/R組細胞內ROS水平、鐵沉積、亞鐵(Fe2+)、MDA水平均升高,過表達DMT1進一步提高了H/ r誘導的ROS水平、鐵沉積、亞鐵(Fe2+)和MDA水平,而敲除DMT1則顯著降低了H/ r誘導的ROS水平、鐵沉積、亞鐵(Fe2+)和MDA水平.
H/R組發現GSH水平和GPX4活性降低,過表達DMT1進一步降低GSH水平,而敲除DMT1則顯著提高GSH水平(圖2l)。
DMT1對GPX4活性無顯著影響(圖2m)。
這些數據表明,DMT1過表達促進H/ r誘導的細胞鐵死亡,而敲除DMT1則顯著抑制H/ r誘導的細胞鐵死亡。
二、HUCB-MSCs和外泌體的分離與鑒定

為了證明hub - mscs衍生的外泌體可以改變心肌細胞的基因表達。證明了外泌體可以被心肌細胞吸收。標記PKH67的hucb - mscs外泌體加入心肌細胞培養液中。孵育6 h后如圖3f所示,大部分心肌細胞獲得了pkh67標記的外泌體,hucb - mscs外泌體在心肌細胞的細胞質中分布相似。
三、hub - msc衍生的外泌體可傳遞miR-23a-3p來抑制ferroptosis

A-B.miR-23a-3p在形成的外泌體中被成功敲低。為了進一步研究外泌體對心肌細胞ferroptosis的影響,構建H/ r誘導的心肌細胞,模擬體外心肌損傷.
C-D. H/ r誘導的細胞中miR-23a-3p表達降低,而與外泌體共培養的心肌細胞中miR-23a-3p表達升高。與hucbmscs -外泌體共培養的心肌細胞中,DMT1 mRNA和蛋白表達與miR-23a-3p相反.
E-M.過表達DMT1可進一步降低H/ r處理心肌細胞的細胞活力和GSH水平,增加細胞凋亡、細胞內ROS水平、鐵沉積、亞鐵(Fe2+)和MDA水平。圖4e -m所示的結果與DMT1表達的變化趨勢相同,說明DMT1是hub - mscs來源的外泌體傳遞miR-23a-3p抑制ferroptosis的關鍵靶點之一。mir -23a-3p耗損HUCB-MSCs外泌體在心肌細胞鐵質下垂中發揮微弱的抑制作用,與NCI-Exo組相比,細胞活力和GSH水平降低,細胞凋亡、細胞內ROS水平、鐵沉積、亞鐵(Fe2+)和MDA水平升高.
N. hub - msc -外泌體對GPX4的活性沒有顯著影響(圖4n)。
總之,hucb - msc衍生的外泌體傳遞miR-23a-3p抑制鐵死亡。
四、hucb - mscs來源的表達mir -23a-3p的外泌體通過靶向DMT1抑制了ferroptosis

A. Starbase軟件預測了miR-23a-3p與DMT1的3 UTR的結合關系。
B. 轉染miR-23a-3p mimic的細胞在DMT1- WT共轉染體系中熒光素酶活性降低,而在DMT1- mut共轉染體系中無明顯變化,說明miR-23a-3p與DMT1直接結合。
C. 用hucb - msc -外泌體孵育時,用過表達的DMT1慢病毒感染心肌細胞。DMT1 mRNA和蛋白表達水平在ExoVector組降低,Exo + DMT1組升高,提示hubmscs -外泌體對DMT1表達的負調控作用(圖5b)。
D. 過表達DMT1慢病毒感染后,hucbmscs -外泌體誘導的細胞活力升高和GSH水平降低,細胞凋亡減少,細胞內ROS水平、鐵沉積、亞鐵(Fe2+)、MDA水平升高,表明hucbmscs來源的表達mir -23a-3p的外泌體通過靶向DMT1抑制了ferroptosis。
五、hucb - mscs -外泌體對AMI小鼠心肌細胞的鐵死亡有抑制作用

E. TTC染色顯示hucb - msc -外泌體減輕AMI小鼠梗死面積,而miR-23a-3pI-Exo減輕AMI小鼠梗死面積的作用明顯減弱.
F. hucbmscs -外泌體給藥可減輕心肌損傷,而miR-23a-3pI-Exo給藥可明顯減弱減輕AMI小鼠心肌損傷的作用。
G. 心肌梗死小鼠心肌組織中tunel陽性細胞比例顯著增加HUC bmscs -外泌體的植入可以消除這種現象。然而,miR-23a-3pI-Exo移植比NCI-Exo移植具有更高的凋亡指數.
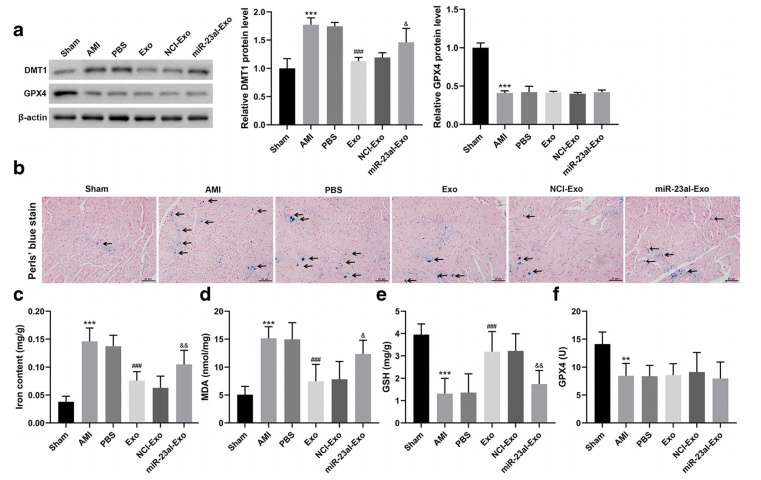
D.HUCB-MSCs外泌體明顯抑制DMT1表達、鐵沉積、亞鐵(Fe2+)和MDA水平,上調GSH水平,但miR-23a-3pI-Exo對這些的作用明顯減弱。
E. HUCB-MSCsexosomes對AMI小鼠GPX4活性沒有顯著影響(圖7f)。這些結果表明HUCB-MSCs外泌體抑制了鐵死亡。