






MGMT基因重排有助于膠質瘤化療耐藥
近月來,一篇名為“ MGMT genomic rearrangementscontribute to chemotherapy resistance in gliomas ”,發表在《Nature Communications》上,由CNIO的Seve Ballesteros基金會腦腫瘤小組負責人MassimoSquatrito博士和北京神經病學研究所的Tao Jiang博士(醫學博士)領導。
“ MGMT啟動子高甲基化是膠質母細胞瘤患者中唯一已知的TMZ反應生物標記。在這里,我們顯示,復發性神經膠質瘤的一個子集攜帶MGMT基因組重排,導致MGMT過表達,與其啟動子甲基化的變化無關。通過利用CRISPR / Cas9技術,我們在神經膠質瘤細胞中產生了一些MGMT重排,并證明MGMT基因組重排在體外和體內均對TMZ產生了抗性。”
研究人員說:“在一組患者中觀察到了MGMT基因的移位。” 這些基因組重排涉及MGMT與其他基因的融合,這意味著MGMT現在受到與其融合的啟動子的調控,這有助于其過度表達。當這種類型的重排發生時,替莫唑胺誘導的DNA損傷得到了非常有效的修復,并且即使在治療下,神經膠質瘤也繼續增長。
為了揭示神經膠質瘤患者的TMZ耐藥情況,研究人員分析了252例經TMZ治療的復發性神經膠質瘤的RNA測序數據,其中新收集了105例(42%)。然后,該團隊整合了臨床信息并進行了生物信息學分析,以確定幾種關鍵改變的突變狀態。CNIO小組使用CRISPR-Cas9在不同的細胞和動物模型中復制了其中的一些易位,并證實它們可以賦予對替莫唑胺的抗性。研究人員補充說:“似乎在原始腫瘤中不存在易位,僅在復發性腫瘤中存在,即在治療原始癌癥后出現的腫瘤。” “這表明抗藥性可能是治療本身的結果。”
他們的發現可能會導致監測治療效果的方法發生變化:“目前,膠質瘤中唯一已知的治療生物標志物是對MGMT啟動子狀態的分析。甲基化后,MGMT基因沉默,預計患者對替莫唑胺有反應。研究表明,當發生基因易位時,該方法不再有效。該啟動子可能仍然被阻斷,但是該基因被其他啟動子過度激活,因此可能導致腫瘤復發。”
研究人員還發現外泌體中存在MGMT易位。研究人員指出,如果這一發現在患者中得到了驗證,那么它將有助于早期發現耐藥性。研究人員的下一個目標是為替莫唑胺耐藥的患者確定新的治療手段。
總而言之,替莫唑胺(TMZ)是一種口服烷基化劑,用于治療膠質母細胞瘤,目前正在成為診斷為高危低級別膠質瘤患者的化療選擇。o -6-甲基鳥嘌呤-DNA甲基轉移酶(MGMT)負責直接修復主要tmz誘導的毒性DNA加合物,即o6 -甲基鳥嘌呤病變。MGMT啟動子高甲基化是目前已知的膠質母細胞瘤患者TMZ反應的唯一生物標志物。此文章表明復發性膠質瘤的一個子集攜帶MGMT基因組重排,導致MGMT過表達,獨立于其啟動子甲基化的變化。通過利用CRISPR/Cas9技術,我們在膠質瘤細胞中產生了一些MGMT重排,并證明MGMT基因組重排在體內和體外都有助于TMZ耐藥。最后,證明了這種融合可以在腫瘤來源的外泌體中檢測到,并可能代表接受TMZ治療的患者中腫瘤復發的早期檢測標志物。
技術路線:

結果:
一、復發性膠質瘤中MGMT基因融合的鑒定

通過分析252例復發性腦膠質瘤的RNA-seq數據,我們在7例患者中確定了8種不同的MGMT融合(約占所有患者的3%,95%可信區間,1.1 5.6%)。值得注意的是,在7例合并MGMT融合的患者中,有6例是女性,這明顯高于預期。重要的是,bootstrapping方法顯示MGMT低甲基化、DNA高突變和MGMT融合之間存在著顯著的相互排他性,表明這些變化在癌癥進展中發揮著替代作用.
A. 總的來說,我們發現38.4%(245例)患者存在IDH1突變,9.4%(23例)患者存在1p/19q共缺失,38%(136例)患者存在MGMT啟動子低甲基化,10.7%(27例)患者存在DNA高突變.
B. 對8種不同的MGMT重排進行了深入研究:HGG中的BTRC-MGMT、CAPZB-MGMT、GLRX3-MGMT、NFYC-MGMT、RPH3A-MGMT和SAR1A-MGMT, LGG中的CTBP2- MGMT和FAM175B-MGMT. 8個伴侶基因中的5個位于染色體10q上,大部分接近MGMT
C. 盡管MGMT融合的左伴子不同,但MGMT轉錄組斷點始終位于MGMT起始密碼子上游12 bp處的MGMT外顯子2的邊界上。在三種重排(SAR1A-MGMT、RPH3A-MGMT和CTBP2-MGMT)中,MGMT編碼序列融合到融合伙伴的5 UTR上。重組嵌合轉錄本發現所有融合體均在框內,MGMT的甲基轉移酶結構域和dna結合結構域均完整,提示MGMT的功能可能保留在融合蛋白中。
D-E我們利用PCR和Sanger測序在有足夠標本的樣本中驗證了基因融合。對于一個患者(CGGA_1729),我們進行了全基因組測序(WGS),該樣本的結構重排分析顯示,大約4.8 Mb的缺失導致FAM175B-MGMT融合.
二、MGMT基因重排導致MGMT過表達

A.為了產生攜帶MGMT融合的細胞系,我們首先將U251和U87細胞,兩種MGMT甲基化的GBM細胞系,與慢病毒載體表達不同的gRNA對組合,指向四種不同的MGMT重排:BTRC-MGMT、NFYC-MGMT、SAR1A-MGMT和CTBP2-MGMT(補充圖2a c)。
通過PCR在基因組水平檢測到預期的染色體重排,并通過Sanger測序證實。然后將新生成的細胞群暴露于TMZ中。存活的克隆只在攜帶不同融合事件的細胞群體中觀察到,而在對照細胞中沒有觀察到(sgCtrl,非靶向sgRNA).
B.TMZ耐藥可能是由于MGMT表達增加導致的,因為重排將MGMT基因置于一個更活躍的啟動子控制之下。實時熒光定量PCR結果顯示,MGMT在攜帶不同融合體的無性系中表達量顯著增加。
與對照細胞相比,MGMT啟動子甲基化狀態沒有變化,甲基化特異性PCR (MSP)證實了這一點。這些結果與在患者隊列中觀察到的結果一致:MGMT重排的患者顯示MGMT表達升高,同時MGMT啟動子甲基化。
使用抗gmt抗體進行Western blot分析,發現MGMT在蛋白水平上明顯過表達,SAR1A-MGMT和CTBP2-MGMT融合克隆尤其明顯(圖2d)。此外,MGMT表達的不同水平可能由參與融合事件的特定基因s啟動子的活性和/或每個特定克隆基因組重排列的拷貝數決定。

為了確定攜帶融合基因的克隆對TMZ的抗性是否由一種全功能MGMT蛋白的過表達決定,而不是由TMZ治療過程中獲得的其他突變引起的,我們分析了O6-芐基鳥嘌呤(O6- bg)對TMZ的敏感性,O6- bg是鳥嘌呤的一種合成衍生物,可抑制MGMT活性。
A.對2個獨立的U251克隆進行克隆實驗,結果表明,與O6-BG共處理后,TMZ的敏感性得以恢復。
B-D.相比之下,錯配修復基因MSH6(一種獨立于MGMT表達的tmz耐藥機制)的細胞敲除在O6-BG存在下也完全耐TMZ。同樣,碘化丙啶染色和EdU摻入分析顯示融合克隆避開了tmz誘導的G2/M期積累,O6-BG共處理能夠重新建立細胞周期停滯。
E-F.高通量顯微鏡定量分析顯示,與sgRNA MSH6細胞相似,TMZ處理未增加γH2AX和53BP1病灶水平,這是DNA雙鏈斷裂細胞特有的DNA損傷標志物.
總之,O6-BG抑制MGMT導致TMZ處理后融合克隆中γH2AX和53BP1焦點的積累。MGMT基因重排誘導的TMZ抗性與MGMT活性存在機制方面的聯系。
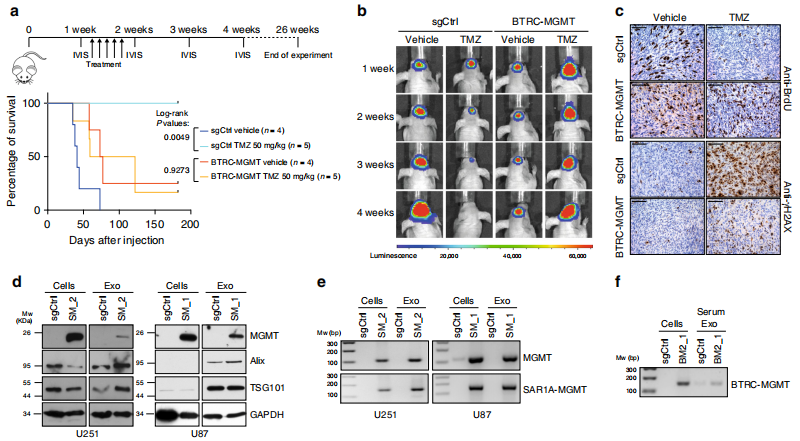
四、MGMT基因融合在體內對TMZ治療有保護作用
通過U251 BTRC-MGMT和對照細胞建立nu/nu小鼠異種移植模型,體內MGMT融合的TMZ耐藥性,之前用表達熒光素酶的構建物轉染。顱內移植后1周,小鼠腹腔注射TMZ (50 mg/ Kg)或DMSO(0.3%) 5天,每周用生物熒光成像(BLI)監測腫瘤生長情況,連續4周。
AMGMT帶有融合腫瘤的小鼠在TMZ組和DMSO組之間沒有顯示出顯著的壽命延長,并且在接受TMZ治療時,與對照組小鼠相比,生存率顯著下降.
TMZ治療顯著延長了sgCtrl轉導h543的小鼠移植后的存活時間,但未能延長表達SAR1A-MGMT重排的小鼠的存活時間(補充圖7h)。BLI分析證實TMZ對正常小鼠的抗腫瘤作用有限.
免疫組化顯示TMZ給藥后,BTRC-MGMT小鼠BrdU摻入增加,γH2AX積累減少.
評估MGMT融合是否可以在EXOs中檢測到。用標準的超離心法從含有SAR1A-MGMT和sgCtrl的條件培養基中純化了EXOs。蛋白質含量的Western blot證實了外泌體特異性標記TSG101和Alix在外泌體中富集. MGMT在表達融合事件的細胞中的存在(圖4d)。
RT-PCR檢測到MGMT融合的mRNA在EXOs中(圖4e)。
原位注射U251 BTRC-MGMT細胞的小鼠血清中分離的外顯子是否也表現出融合轉錄。值得注意的是,RTPCR分析證實了btrc - mgmt衍生的循環血液外顯子中存在cDNA融合片段.