






單細胞測序與TCGA數據助力膀胱癌發表高分文章
膀胱癌是世界上最普遍的泌尿生殖系統惡性疾病之一,每年診斷出約430,000例新病例,造成165,000多例死亡。盡管在癌癥生物學和治療方面已取得實質性進展,但膀胱癌患者的臨床結果仍不令人滿意。在過去的十年中,與藥物發現的治療靶標有關的腫瘤微環境(TME)一直是癌癥生物學研究的熱門領域。值得注意的是,膀胱癌是免疫力最低的浸潤癌之一,這可能是對抗PD1治療反應差的原因,為膀胱癌設計新的治療策略一直是一項艱巨的任務。之前,已經報道了膀胱癌的分子亞型顯示出不同的細胞類型特異性表達模式,這表明膀胱癌的異質性至少部分是由微環境內部的不同細胞類型組成的。但是,直到最近,相關的研究還很少。近期,華中科技大學同濟醫學院附屬醫院泌尿科陳科教授及其團隊分析了膀胱尿路上皮癌或癌旁粘膜樣品中單細胞的轉錄組,并繪制了膀胱癌組織內整個TME的圖集,促進了對膀胱癌患者之間異質性的理解,并為膀胱尿路上皮癌的個體化治療提供了基礎。相關研究以“Single-cell RNA sequencing highlights the role of in?ammatory cancer-associated ?broblasts in bladder urothelial carcinoma”為題發表在Nature communications雜志上,雜志影響因子12.121。
技術路線:
結果:
1、膀胱癌細胞異質性
作者通過質量控制消除了批次之間的批次效應,將52721個單細胞聚集為八個主要簇,簇特異性基因被用來標記來注釋細胞類型:上皮(EPCAM+)細胞;內皮(CD31+)細胞;兩種類型的成纖維細胞(COL1A1+)-iCAF(PDGFRA+)和肌CAF(mCAFs)(RGS5+);B細胞(CD79A+);骨髓細胞(LYZ+);T細胞(CD3D+);和肥大細胞(TPSAB1+)(a,b)。隨后,將EPCAM +上皮細胞(EPC)重新聚集以產生17個簇,通過InferCNV證實了癌細胞顯示出患者特異性的表達模式可能是由拷貝數變異(CNV)引起的極高的異質性的假設,且發現腫瘤組織中的一些細胞幾乎不具有CNV,表現出與正常EPC相似的表達模式,同時發現一系列基因在對照組和CNV未檢出組中特別表達,但在腫瘤細胞中幾乎不存在(c,d)。基因本體富集分析表明,這些基因富集于免疫相關途徑,特別是B細胞相關途徑(e)。與正常上皮細胞相比,癌細胞幾乎喪失了產生免疫球蛋白的能力,免疫組化分析發現它們表達的MHC-II分子水平較低(f,g),表明膀胱癌細胞可能下調免疫原性以逃避免疫檢測。通過基因組變異分析(GSVA)進行的途徑分析顯示,CNV高水平人群中E2F靶標、MYC靶標和G2M檢查點途徑富集,而炎癥和其他免疫相關途徑則被下調(h),進一步證實了處于膀胱癌晚期的癌細胞下調了免疫原性并顯示出高增殖能力。

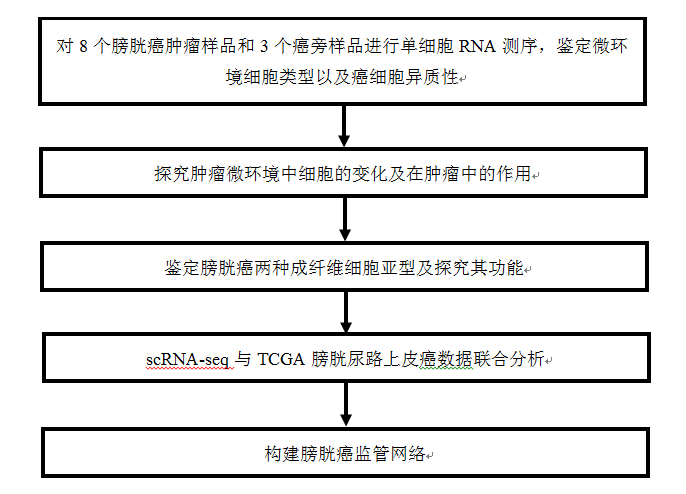
2、腫瘤區域的單核細胞經歷M2極化
作者共鑒定出7種細胞類型:腫瘤相關的巨噬細胞、CD1C+樹突狀細胞、單核細胞、增殖性骨髓細胞、交叉呈現DC、卵泡B細胞和LAMP3+DC。兩個細胞簇均表達髓樣標志物以及上皮或內皮標志物被認為是雙峰(a)。單核細胞大部分起源于正常的粘膜組織,而腫瘤相關巨噬細胞則富集在膀胱癌組織中。此外,這兩種細胞類型的轉錄組表現出連續的變化,表明募集到腫瘤區域的單核細胞被重新編程為腫瘤相關巨噬細胞(b)。作者發現BACH1,MAFG和NFE2這三個基序的活性被下調,而MAF,STAT1和STAT2基序的激活導致M2極化過程,為抑制或逆轉免疫抑制微環境的形成提供了潛在的目標(c,d)。此外,在這種分化過程中,共抑制因子CD274,LGALS9,CD276,TIGIT和PDCD1LG2均被上調,而共激活因子被下調(e)。三個DC亞組中,LAMP3+ DC組表達了各種編碼細胞因子的基因,包括CCL17,CCL19和CCL22(f),這些細胞因子幾乎完全來源于膀胱癌的LAMP3+ DC(g)。LAMP3+ DCs顯示出最高水平的CD274(f),甚至高于從膀胱癌組織的Tregs中觀察到的水平,表明該DC亞組可以直接抑制CD8+ T細胞或通過募集Tregs進入腫瘤區域。在TCGA膀胱尿路上皮癌隊列中,LAMP3+ DC簽名與均為CCR4+的Treg簽名和Th2簽名高度正相關,但與CTL簽名沒有高度相關(h)。
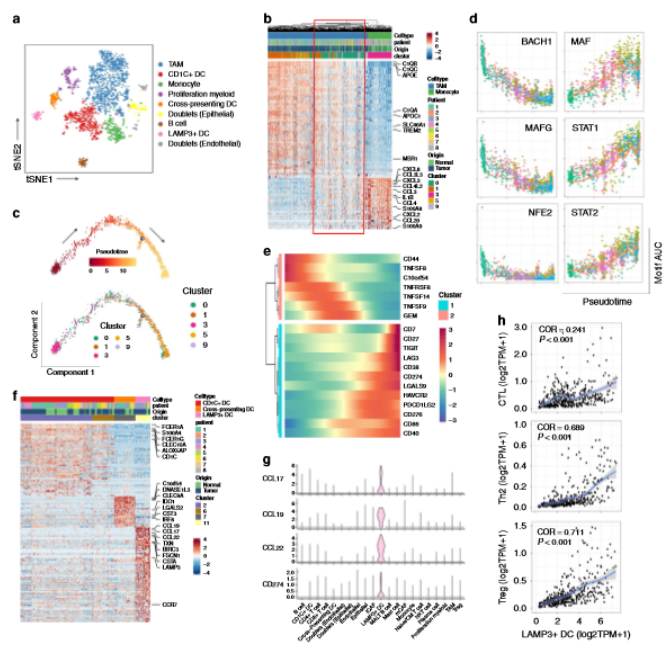
3、膀胱癌成纖維細胞亞型
成纖維細胞(COL1A1+)分為兩種不同類型:PDGFRA+成纖維細胞表現出各種細胞因子和趨化因子的強表達,包括CXCL12,IL6,CXCL14,CXCL1和CXCL2;RGS5+成纖維細胞具有與肌癌相關的成纖維細胞(mCAFs)相似的特征(a,b)。通過免疫熒光法評估了腫瘤和非惡性膀胱基質組織中現有的iCAF和mCAF發現在,不同類型的癌癥中CAFs具有類似的亞群(c)。為了調查每個亞組的功能,作者對iCAF和mCAF的差異表達基因進行了GO富集分析發現iCAF與細胞外基質的組織,細胞遷移的調節和血管生成有關,而肌肉系統過程,粘著斑和細胞外基質相關的途徑則明顯富含mCAF(d)。GSEA同樣顯示iCAF與細胞外基質降解有關,表明其在腫瘤轉移中的潛在作用。細胞因子-細胞因子受體的相互作用途徑也豐富了iCAFs。相反,肌肉收縮和PGC1A途徑富含mCAF(e,f)。由于細胞因子與細胞因子受體的相互作用在iCAF中富集,因此研究了膀胱癌TME中細胞因子的表達水平。CXCL12與TCGA膀胱尿路上皮癌隊列中的腫瘤相關巨噬細胞簽名呈正相關,而較高水平的CXCL12與不良預后顯著相關。免疫熒光實驗發現CXCL12在膀胱癌組織中通過iCAFs表達(g-h)。

4、iCAF的增殖作用
通過SCENIC分析確定了兩個CAF亞組中的必需基序,TCF21和TWIST2基序在iCAF中被高度激活(a,b)。隨后,通過GO富集預測iCAF具有生長因子活性,并分析了膀胱癌TME中VEGF,FGF和IGF家族的表達水平(c),在這些生長因子中,IGF1是iCAF特異性的組因子,與總體生存期較差有關(d,e)。為了驗證其促增殖作用,作者通過流式細胞術對iCAF進行了分類,并將它們與膀胱尿路上皮癌細胞系進行了共培養,顯示出更高的增殖能力,這證實了iCAF在膀胱癌中的腫瘤作用(f,g)。
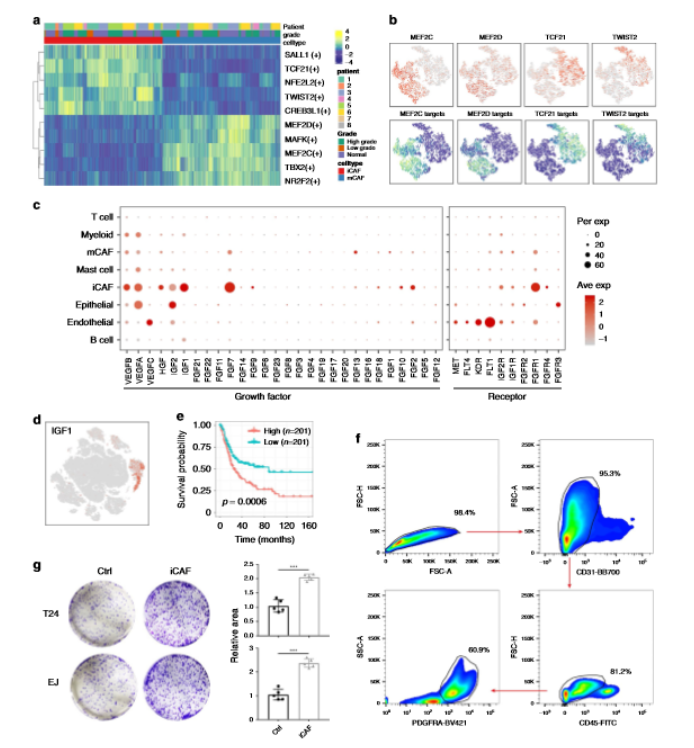
5、scRNA-seq與公共數據集相關分析
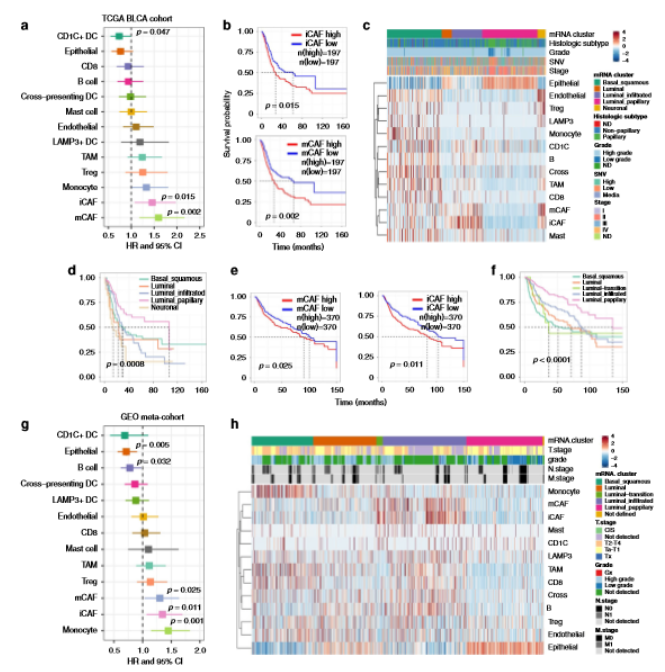
為了研究本研究中確定的細胞類型的臨床作用,作者使用CIBERSORTx 評估TCGA膀胱尿路上皮癌隊列樣本中每種細胞類型的比例,發現只有iCAF和mCAF的積累與較差的總體生存率相關(a,b)。當將細胞分數數據與臨床信息相關聯時發現先前描述的分子亞型之間的細胞類型豐度變化很大,腫瘤純度最高的乳頭狀乳頭狀瘤的預后最好,而其他四組的總生存率則無明顯差異,其中基底鱗癌顯示出最低的腫瘤純度,還富集了T細胞,表明抗免疫檢查點療法可能適合這些患者(c,d)。隨后,作者擴大了臨床隊列,從GEO和ArrayExpress數據庫中收集了3000多個微陣列分析的膀胱癌和非惡性粘膜樣品,通過Combat功能消除了可能的批量效應,然后使用ConsensusClusterPlus將2959個腫瘤樣品分為五個主要簇,四個主要的團簇顯示了與熒光團簇相似的特征:乳頭狀團簇、管狀團簇、熒光浸潤團簇和基底鱗狀團簇。另一簇與TCGA 膀胱尿路上皮癌中的神經組不同,同時具有光鱗狀和管腔特征,故命名為管腔躍遷。該薈萃隊列中的總生存率與TCGA高度對應,證明了CAF在膀胱癌進程中的重要作用(e-f)。
6、構建基于iCAF的監管網絡
作者通過CellphoneDB2研究了細胞類型之間的細胞-細胞相互作用網絡,iCAFs與其他細胞類型的相互作用最多,并且與ECs的相互作用特別強(a)。iCAFs中CXCL12表達水平較高,其受體包括DPP4、CXCR3、CXCR4和ACKR3 (CXCR7)。由于CXCR4和CXCR3在免疫細胞上廣泛表達,iCAFs分泌CXCL12負責膀胱癌的免疫浸潤狀態。iCAFs分泌的CCL2和CXCL1可與ECs表面高表達的ACKR1相互作用。這些細胞因子之前被報道與膀胱癌的轉移有關,確定它們的起源為iCAFs(b,d)。作者認為,iCAFs可產生VEGF,包括VEGFA和VEGFB,它們與內皮細胞上的VEGF受體(FLT1、KDR、MET和FLT4)結合,促進血管生成。此外,FGFR1在iCAFs和ECs上表達,而FGFR3在腫瘤細胞上表達。這些受體可與FGF結合,包括來自iCAFs的FGF2和FGF7,并表現出促增殖作用。IGF1R是IGF1的受體,在腫瘤細胞和間質細胞上均有表達,提示iCAF也可介導對順鉑的耐藥。值得注意的是,腫瘤細胞中VEGFA表達水平較高,其促血管生成能力最強。此外,高級別腫瘤細胞在iCAFs上高表達IGF2,并與IGF2R相互作用,促進腫瘤進展(c, e)。綜上所述, iCAFs可以促進腫瘤細胞和基質細胞的增殖。
結論:
1、作者在膀胱癌微環境中鑒定出19種不同的細胞類型,表明腫瘤內異質性很高。
2、研究發現腫瘤細胞下調了MHC-II分子,提示癌細胞的下調免疫原性可能有助于形成免疫抑制性微環境,且發現單核細胞在腫瘤區域經歷M2極化并分化。
3、LAMP3+ DC亞組可能能夠募集調節性T細胞,可能參與免疫抑制性TME的形成。
4、炎癥相關的成纖維細胞與腫瘤不良的預后顯著相關。
參考文獻:
Chen, ZH; Zhou, LJ; Liu, LL,et al. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nature communications. 2020.