上皮-間質轉化影響頭頸癌中鐵死亡
研究背景:鐵死亡是一種鐵依賴的脂質過氧化引發的新型細胞死亡方式。鐵死亡是一種抑制抵抗癌癥治療的治療方法,具有上皮-間充質轉化(EMT)特性。EMT是上皮細胞打破細胞間和細胞間基質黏附狀態的重要過程,賦予癌細胞化療耐藥性。因此,Jaewang Lee等人研究了EMT表觀遺傳重編程在促進頭頸癌(HNC)細胞的鐵死亡中的治療潛力。(Redox Biology IF= 9.986)
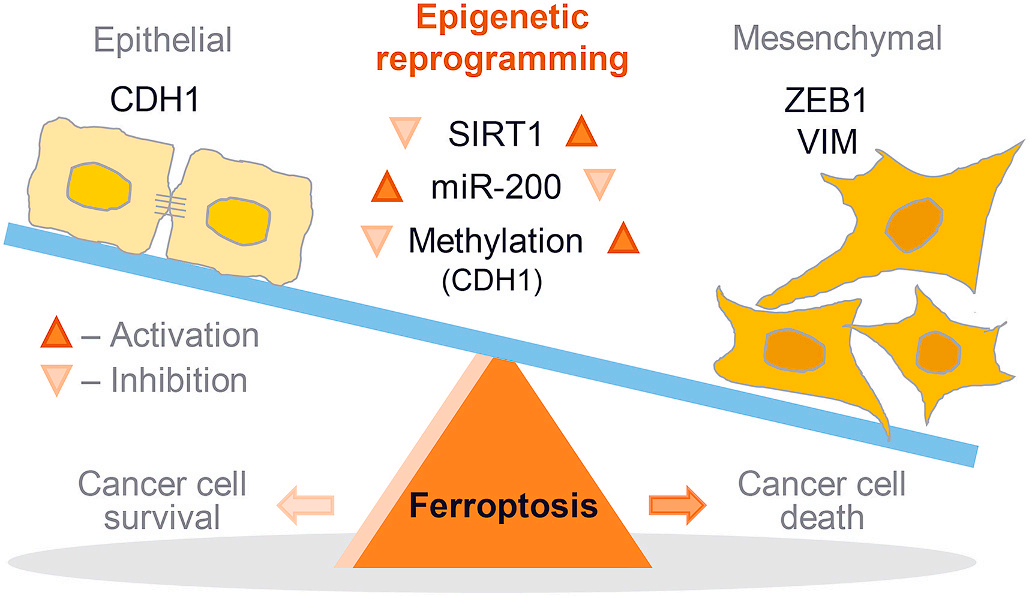
技術路線圖:
研究結果:
1.細胞密度和EMT標記物與鐵死亡敏感性相關
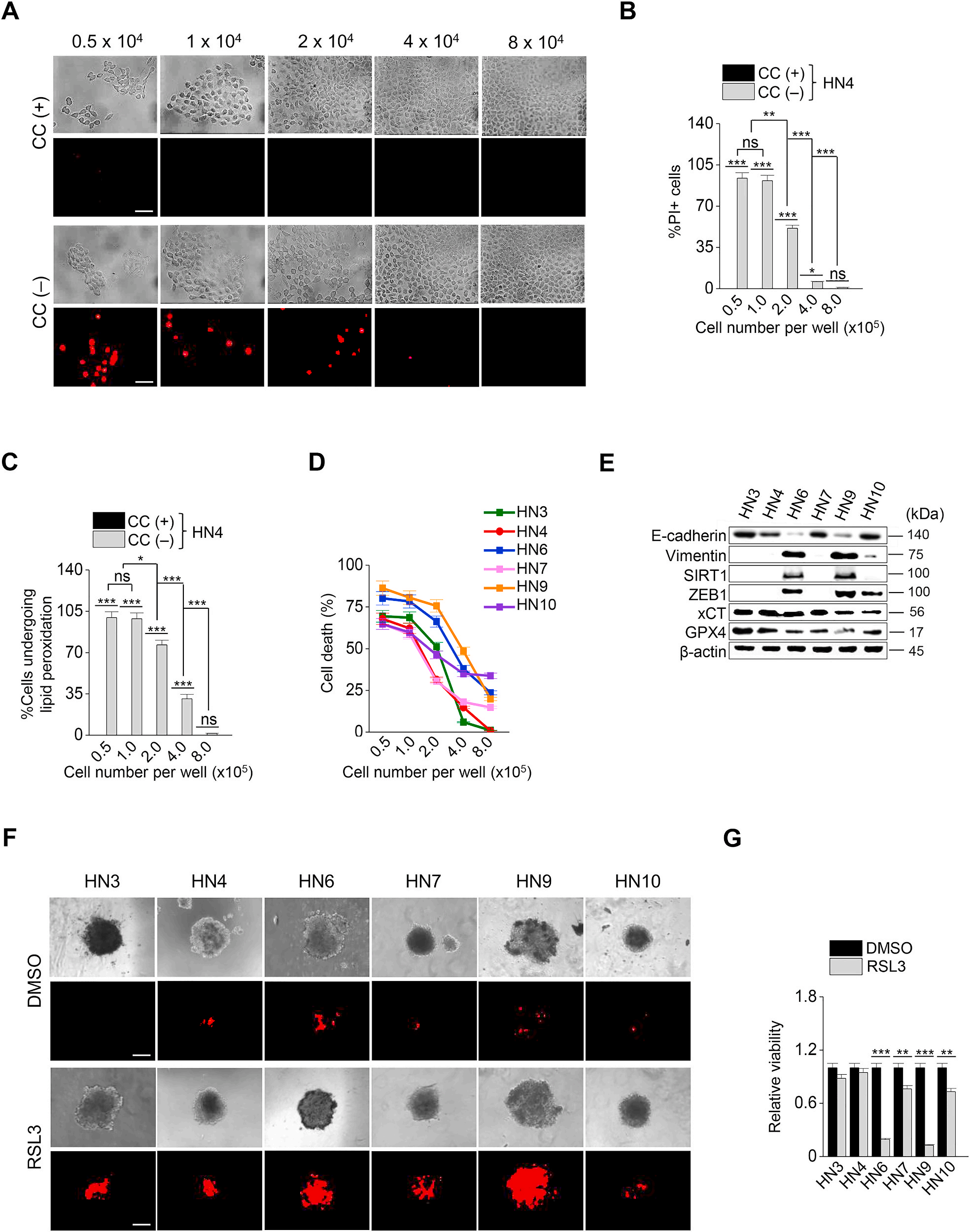
首先在胱氨酸耗竭培養條件下進行細胞死亡試驗,確定細胞密度是否會影響鐵死亡的敏感。在HN4癌細胞中,PI陽性細胞占比與每孔細胞數負相關(圖1A和B)。細胞脂質過氧化水平也隨著HN4的增加而降低(圖1 C)。結果表明,細胞密度與鐵死亡敏感性有關。檢測了6個HNC細胞系的細胞死亡證實,癌細胞密度越大,存活時間越長 (圖1D)。E-cadherin上皮標志物在HN3、HN4、HN7和HN10中高表達,而間質標志物vimentin和ZEB1在HN6和HN9中高表達(圖1E)。結果表明,EMT標記物的表達水平與鐵死亡的敏感性相關(圖1D和E)。在具有間質性特征的HN6和HN9細胞中,鐵溶性誘導物RSL3暴露導致的細胞死亡顯著高于其他具有上皮性特征的HNC細胞系 (圖1F和G)。綜上所述,這些數據表明向間充質性質的轉變可能使癌細胞更容易受到鐵胞作用的影響。
Figure1
2. E-cadherin調節鐵死亡的誘導
轉化生長因子(TGF-β)處理HN3和HN4細胞,誘導EMT。沉默CDH1誘導了ZEB1和vimentin的表達(圖2A)。RSL3或磺胺嘧啶處理時,沉默CDH1使細胞更容易受到鐵死亡誘導劑的誘導。CDH1沉默增強了細胞死亡的增加,這與暴露于RSL3或磺胺嘧啶的癌細胞中不穩定鐵水平的增加是同時發生的(圖2D)。CDH1過表達降低了ZEB1和vimentin在HN6細胞中的表達(圖2G)。RSL3、磺胺嘧啶或胱氨酸耗竭處理HN6細胞時,4-HNE和PTGS2表達增加,但在過表達CDH1的細胞中,增加的表達減少(圖2H)。過表達CDH1組細胞死亡降低 (圖2I);細胞內不穩定鐵,脂質過氧化,ROS生成的增加明顯降低(圖2 J K L); NAD/NADH比值下降 (圖2N);而GSH水平升高(圖2M)。這表明,在HN6和HN9中過表達CDH1可能增強具有間充質特性的癌細胞的抗氧化系統。采用小鼠模型移植顯示,CDH1過表達時,腫瘤體積的減少程度低于經磺胺嘧啶治療后的載體對照(圖2O)。磺胺嘧啶處理后GSH含量降低,但NAD/NADH比值增加(圖2P Q)。綜上所述,結果表明控制E-cadherin在癌細胞中的表達可以調節鐵死亡的易感性。
Figure 2

3. ZEB1的表達改變了癌細胞對鐵死亡的敏感性
在HN6和HN9細胞中,沉默ZEB1誘導E-cadherin表達,抑制vimentin表達(圖3A)。暴露于RSL3或磺胺嘧啶后,細胞活力減少 (圖3B)。當使用RSL3、磺胺嘧啶或胱氨酸耗竭時,ZEB1沉默的癌細胞中LIP、脂質過氧化和ROS的增加也低于對照組 (圖3C-E)。ZEB1在HN3和HN4細胞中過表達,誘導vimentin表達并抑制E-cadherin(圖3F)。RSL3、磺胺嘧啶或胱氨酸耗竭處理HN4細胞時,4-HNE和PTGS2表達增加,但在過表達ZEB1的細胞中,表達增加顯著(圖3G)。過表達ZEB1,RSL3或磺胺嘧啶處理的pi陽性細胞明顯增多,細胞存活率明顯下降 (圖3H、圖I)。LIP、脂質ROS和胞漿內ROS的數量,NAD/NADH比值顯著增加 (圖3J L N)。GSH水平降低(圖3M)。體內實驗結果顯示,過表達ZEB1組的腫瘤體積更小(圖3O)。過表達ZEB1組,磺胺嘧啶處理后GSH含量下降,NAD/NADH比值升高(圖3P和Q)。綜上所述,結果表明,腫瘤細胞中ZEB1的調控能夠調節鐵死亡的易感性。
Figure 3
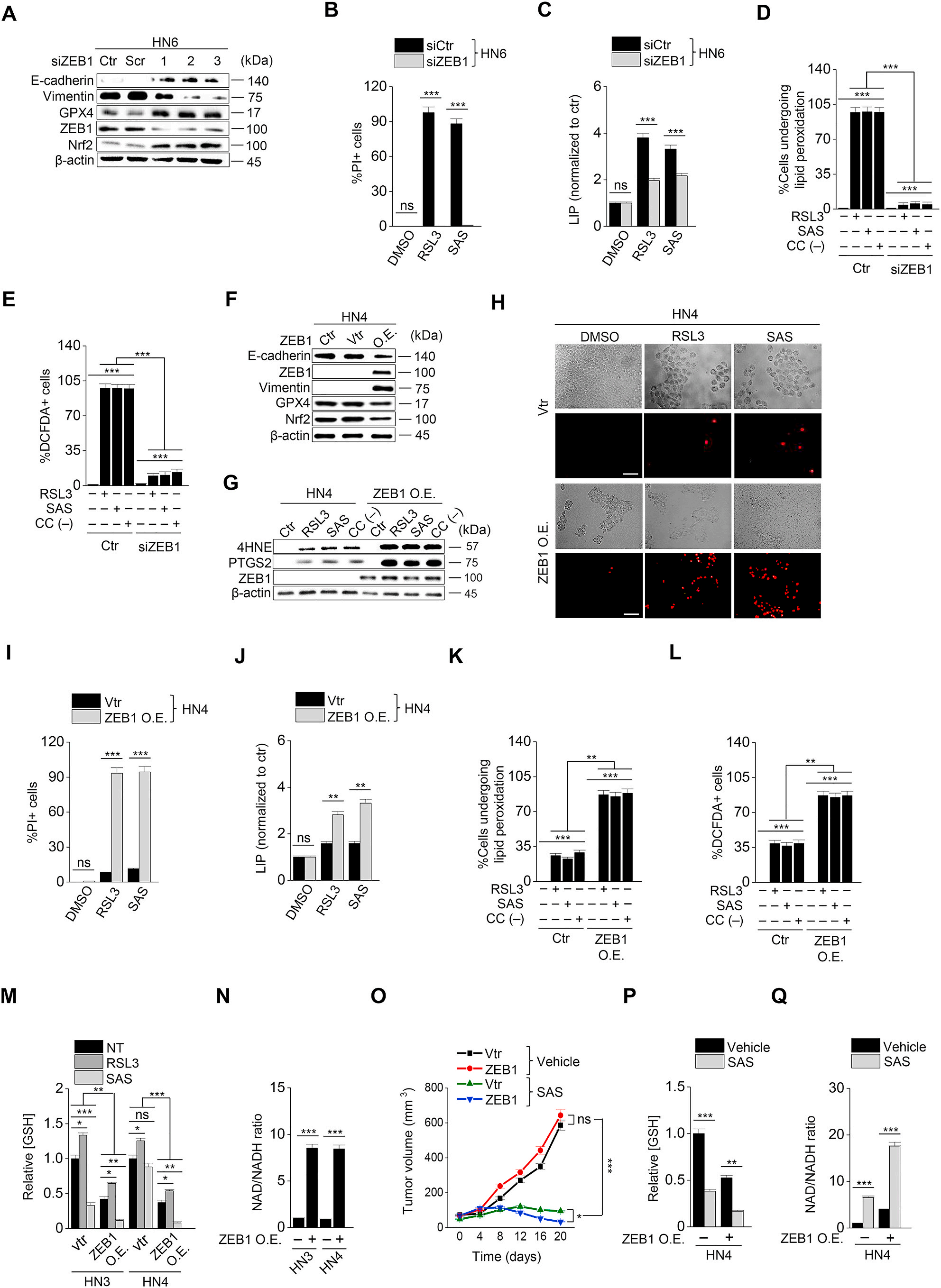
4. SIRT1的激活促進HNC細胞的鐵死亡
探討針對ZEB1的EMT的表觀遺傳重編程是否能夠調節鐵死亡。SIRT1通過激活ZEB1的功能調節EMT。在HN6和HN9癌細胞中, SIRT1沉默或Ex-527(SIRT1特異性抑制劑)處理降低了ZEB1和vimentin的表達,但增加了E-cadherin的表達(圖4A)。與暴露于RSL3或磺胺嘧啶的對照組相比,SIRT1沉默或Ex-527治療后的癌細胞細胞存活率顯著提高,細胞死亡減少 (圖4B、C)。此外,不穩定鐵、脂質ROS和胞漿內ROS的增加比對照組細胞少(圖4D F)。然而,SIRT1誘導劑白藜蘆醇顯著降低RSL3或磺胺嘧啶處理的HN3和HN4的存活率(圖4 G)。在HN3和HN4中,RSL3或磺胺嘧啶與SIRT1特異性誘導劑SRT1720的組合比增加組合基質的對照更顯著地降低了細胞存活率,特別是在高濃度組合下(圖4H和I)。SIRT1720或白藜蘆醇處理增加了ZEB1和vimentin的表達,但降低了E-cadherin的表達(圖4J)。綜上所述,這一結果表明,SIRT1在HNC癌細胞中的控制可能通過抗氧化能力減弱而影響鐵死亡的誘導作用。
Figure 4
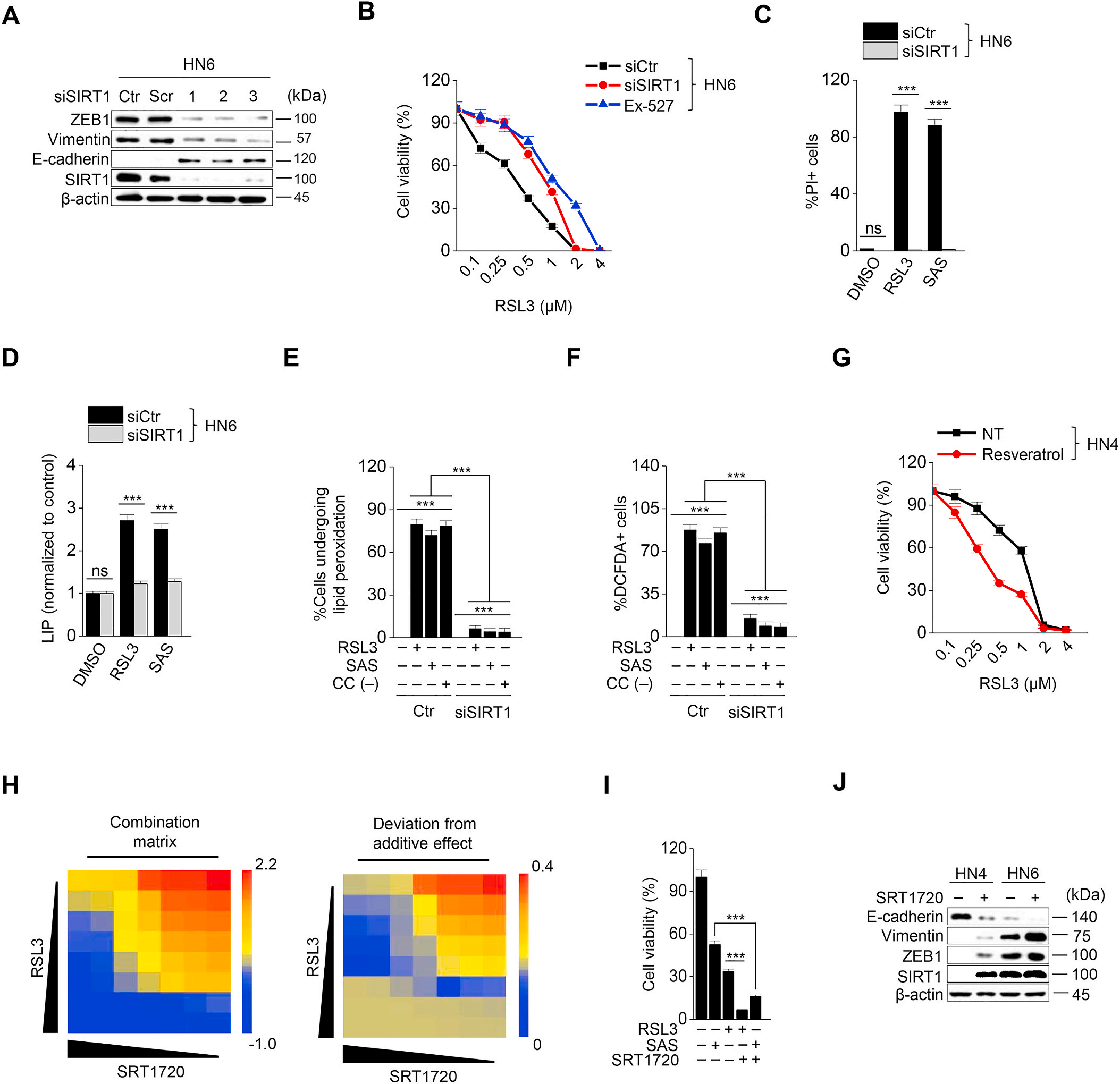
miR-200家族通過抑制ZEB1抑制EMT。首先轉染miR-200家族抑制劑(圖5A),然后在HNC細胞中檢測RSL3、磺胺拉嗪或胱氨酸耗竭后細胞存活率、LIP和ROS的生成。MiR-200家族抑制劑增加了ZEB1和vimentin的表達,但降低了E-cadherin的表達(圖5B)。mir -200a抑制細胞中,HN3和HN4的細胞活力顯著下降,而pi陽性細胞比例的增加多于對照組(圖5 C和D)。與對照組相比,使用miR-200a inhibitor的細胞內不穩定鐵、脂質過氧化和細胞內ROS水平顯著升高(圖5 E G)。結果表明, miR-200家族抑制劑在癌細胞中誘導的EMT可能增強了對鐵死亡的敏感性。
Figure 5

6. 5-Azacitidine轉變為上皮性質,減弱鐵死亡敏感性
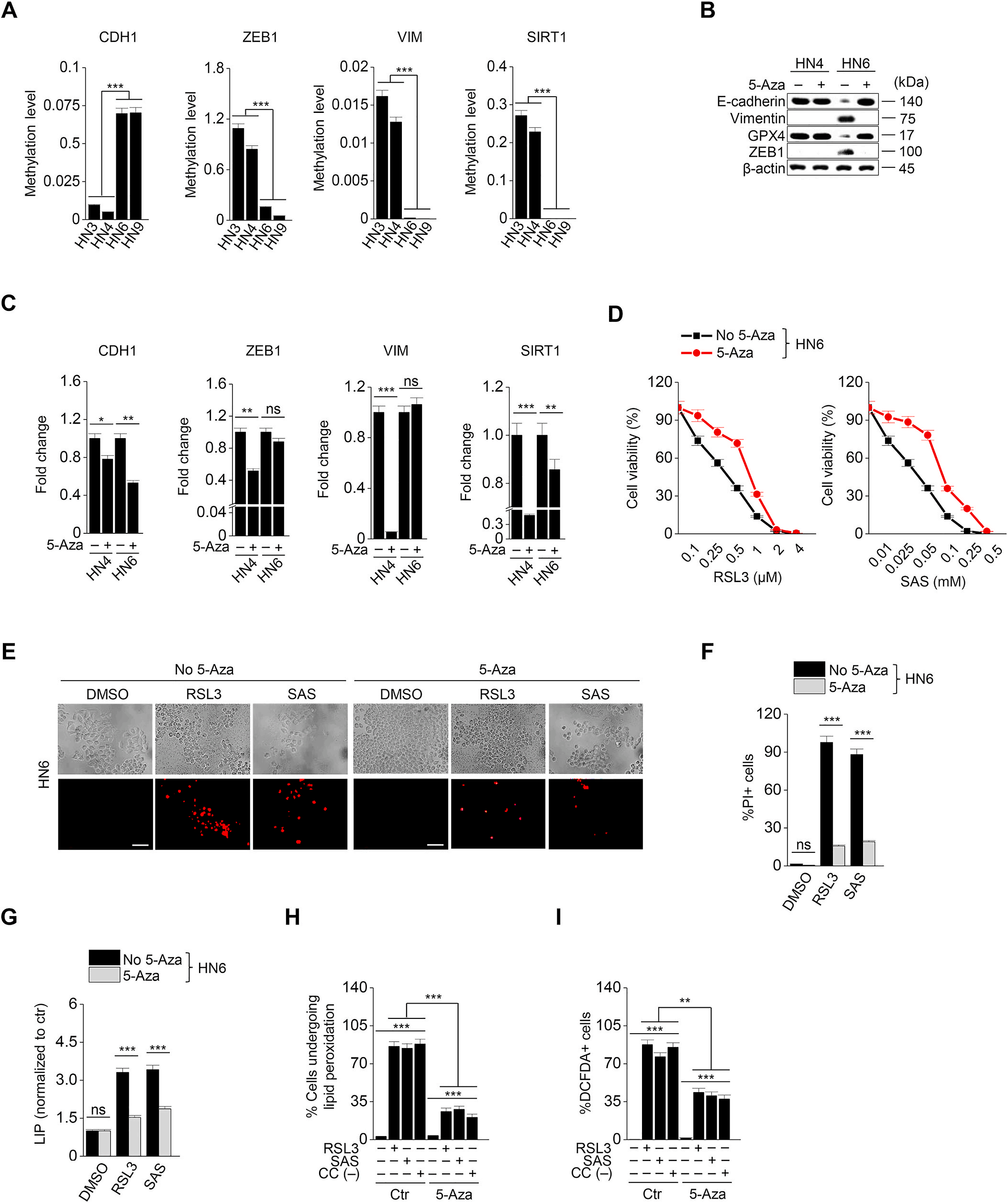
EMT受表觀遺傳學調控,文章中確認了EMT相關基因的甲基化水平。基因的甲基化水平與蛋白質的表達相反,如圖1E所示。CDH1的甲基化水平在間質性的HN6和HN9中較高,而ZEB1、VIM和SIRT1的甲基化水平在上皮性的HN3和HN4中較高(圖6A)。5-Azacitidine是一種去甲基化劑,已知可以預防EMT。5-Azacitidine用RSL3或柳氮磺吡啶處理HN4和HN6。5-azacitidine治療誘導HN6細胞中CDH1甲基化水平降低和ZEB1,波形蛋白表達增加 (圖6 B和C)。這也導致了RSL3或磺胺嘧啶治療后的鐵死亡減少(圖6 D F)。在HN6細胞中,RSL3, 柳氮磺胺吡啶和胱氨酸耗竭引起的不穩定鐵, 脂質過氧化, 胞質ROS的增加被5-Azacitidine減弱(圖6G I)。然而,細胞死亡和存活率、不穩定鐵和脂質過氧化水平在HN4之間無顯著差異。表明5-Azacitidine可能通過去甲基化上皮標志物CDH1來降低鐵亡誘導劑的敏感性。
Figure 6