






NLRP3和鈣離子積累共同推進類風濕性關節炎進程
增加細胞外Ca2+濃度([Ca2+] ex)在單核細胞內通過鈣離子敏感受體(CaSR)驅動激活NLRP3炎癥小體。但是在類風濕性關節炎中的具體機制仍不清楚,本文對該機制進行了深入探討,并挖掘出重要的代謝通路。本文在2020年8月25日發表在《Nature Communications》期刊上,其影響因子12.121。
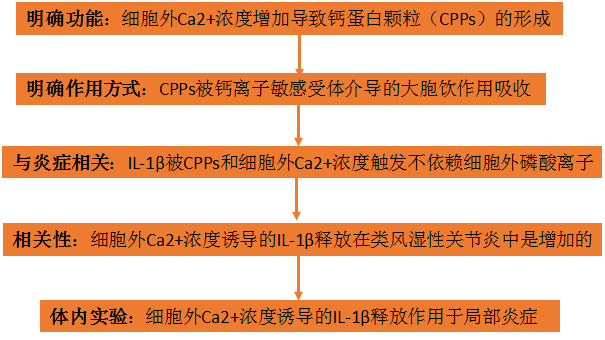
研究結果:
1、[Ca2+] ex的增加導致鈣蛋白顆粒(CPPs)的形成
為了探究鈣離子敏感受體(CaSR)驅動的骨髓細胞內導致NLRP3依賴的IL-1β產量,構建了CaSR敲除的單核細胞THP-1細胞系。如Fig. 1a所示,[Ca2+] ex誘導的IL-1β響應在CaSR敲除的單核細胞THP-1細胞中被顯著抑制,而對ATP和尿酸一鈉(MSU)晶體的響應不受影響。用小鼠外周血單核細胞進行骨髓特異性CaSR敲除(B6.129P2-Lyz2tm1(cre)Ifo7jx-CaSRΔflox/Δflox)實驗證實了這種CaSR效應,研究發現,在5.6mM [Pi]ex存在的情況下,經[Ca2+]ex刺激后,IL-1β分泌物分泌顯著減少(Fig.1b)。
使用含有1-5.6mM的Pi的RPMI1640培養基,研究Pix對[Ca2 +] ex誘導的單核細胞中IL-1β釋放的影響,其中加入[Ca2 +]作為氯化鈣作用。在≥3mM的Piex下,[Ca2 +] ex觸發的IL-1β釋放增加具有濃度依賴性(Fig.1c)。在低Piex下沒有[Ca2 +] ex效應,而且高Piex與低[Ca2 +] ex結合也不會誘導IL-1β釋放(Fig.1c)。如Fig.1d所示,CPPs只能在≥3mM的Pix情況下檢測到,且在沒有[Ca2 +]的情況下不形成鈣離子顆粒。孵育20小時后,CPPs的大小僅略有增加,與顆粒形成并行,由于Ca 2+在顆粒中的結合,[Ca 2+]逐漸降低,通過此過程,添加的Ca2 +會在20h內完全用完(Fig.1e)。WB可以檢測到CPPs中確實存在胎球蛋白(Fig.1f),其顆粒形態為非晶體的,無固定性狀的,膠體結構(Fig.1g和h)。電子顯微鏡與能量色散x射線能譜(EDX)結合揭示在最初的4小時內,CPPs鈣和磷的含量大致相等。

Fig. 1 In the presence of [Pi] and fetuin-A, addition of [Ca2+] triggers calciprotein particle formation
為了證實被[Ca2+]刺激的單核細胞中CPPs的存在,使用TEM和掃描透射電鏡(STEM),可以看到靠近細胞表面的細胞外間隙中有電子致密顆粒,胞漿中也有(Fig. 2a, b)。顆粒的大小和元素組成與FBS補充的2.5mM [Ca2 +]的RPMI中檢測到的CPP相一致,EDX分析表明,這些顆粒含有大量的Ca2 +,表明它們的大小和組成與上述CPP確實相同(Fig.2c,d)。

Fig. 2 Uptake and elemental composition of intracellular calciprotein particles
2、CPPs被CaSR介導的大胞飲作用吸收
增加[Ca2 +] ex刺激大胞飲作用,所以作者猜測自發形成的CPP可能被單核細胞吞噬由于[Ca2 +] ex誘導的CaSR信號,于是作者使用流式細胞術檢測單核細胞的大胞飲作用。如Fig.3a, b所示,單核細胞的大胞飲作用是以一種劑量依賴的方式受到[Ca2 +] ex調節,而不是依賴于[Pi]ex。此外,[Ca2 +] ex誘導的鈣離子大胞飲吸收是一個主動耗能的過程,其37℃時的效果強烈,4℃時則被抑制,并且該過程可以通過細胞松弛素D或latrunculin A(兩種肌動蛋白聚合抑制劑)預處理廢除掉(Fig.3c-e)。鑒于Calhex231和NPS2143,兩種CaSR的特異性負變構調節器可以抑制單核細胞大胞飲作用,證實了[Ca2 +] ex誘導的單核細胞大胞飲作用確實是受到CaSR信號驅動(Fig.3f)。
為了確認[Ca2 +] ex誘導的胞內CPPs的攝取用共焦拉曼顯微鏡(CRM)對單核細胞進行了實時成像(CRM, Fig. 3g, h)。CPPs在無細胞介質中的拉曼光譜(Fig. 3h,左)顯示了與羥基磷灰石晶體顆粒高度相似的指紋圖譜。孵育60min后,CPPs在細胞膜附近被發現,并內化在單核細胞中(Fig.3g)。從胞內CPPs提取的拉曼光譜(Fig. 3h,右)顯示胞內PO4 - 3振動存在對稱的拉伸和彎曲模式。這些都證實了單核細胞中[Ca2 +] ex誘導的CPPs的被攝取進入細胞。
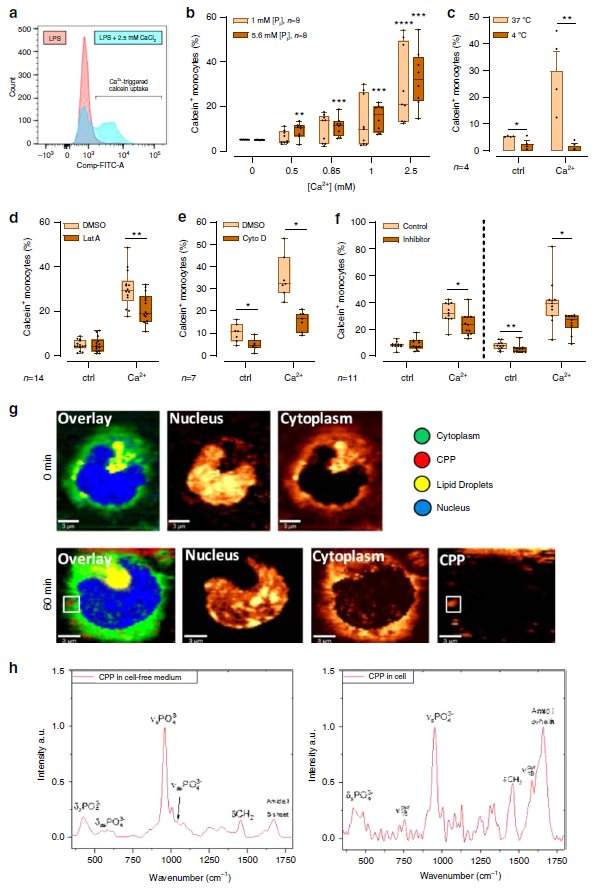
Fig. 3 [Ca2+] ex induces macropinocytosis
接下來,使用流式成像觀察大胞飲作用吸收CPPs通過將單核細胞與結合Ca2+的熒光染料共孵育。熒光圖中細胞內鈣離子信號即表示CPPs的積累。如Fig.4a所示,2.5mM [Ca2+]ex驅動單核細胞對CPPs的吸收,若沒有[Ca2+]ex則吸收量極少。當使用另一種CaSR級聯的物質鋇代替[Ca2+]ex的時候也能觀察到類似的CPPs吸收(Fig. 4b)。
[Ca2+]ex誘導的和[Pi]ex誘導的響應曲線顯示對照組細胞中添加1mM [Ca2+]時有快速的細胞反應,在CaSR缺乏的THP-1細胞中響應顯著降低(Fig. 4d)。然而,CaSR缺乏的細胞仍然對[Ca2+]的添加表現出一些反應,很可能是由于CaSR的獨立作用。[Ca2+]ex誘導的和[Pi]ex誘導的動態質量重新分配(DMR)響應顯示Ca2+加入后的30min開始二次緩慢增加,單核細胞DMR對增加的[Ca2+]ex的響應受到[Pi]ex的與濃度相關的信號增強的影響(Fig. 4e)。

Fig. 4 Macropinocytosis of CPPs depends on CaSR signaling.
3、IL-1β被CPPs和[Ca2+]ex 觸發是不依賴[Pi]ex的
為了探究CPPs在[Ca2+]ex 誘導的NLRP3炎癥小體激活中的作用,在低濃度[Pi]培養基中,在預先形成的CPPs存在的情況下,用[Ca2+]ex刺激單核細胞,以排除從頭形成CPPs的可能性。在2.5mM [Ca2+]ex存在的情況下結果發現CPPs可觸發濃度依賴性IL-1釋放,如果沒有進一步的Ca2+添,CPPs對IL-1β釋放的影響很小(Fig. 5a)。單核細胞[Ca2+]誘導CPP攝取后IL-1β釋放的結果顯示,在有[Ca2+]ex存在的低[Pi]介質中,CPP的形成在3小時后就開始了,并且比在有5.6 mM [Pi]存在的介質中生產[Ca2+]的速度更快(Fig.5b)。Fig. c結果表明[Ca2+]ex 誘導的IL-1β釋放依賴于肌動蛋白聚合。CA-074-Me的藥物組織蛋白酶B抑制降低了[Ca2+]誘導的IL-1β抗氧化反應,該結果進一步支持了被吞噬的CPPs的溶酶體消化有助于[Ca2+]ex誘導的炎癥小體激活和單核細胞中IL-1β釋放(Fig. 5d)。過濾掉大顆粒CPPs后,[Ca2+]ex誘導的IL-1β釋放顯著下降,說明CPPs的大小和濃度與炎癥小體的激活有關(Fig. 5e)。當通過增加培養基中的胎球蛋白-A濃度來增加顆粒生成過程中胎球蛋白-A和[Ca2 +]的比例時,觀察到濃度依賴性的IL-1β釋放降低(圖5f)。 添加PFA抑制了[Ca2 +]濃度依賴誘導的IL-1β釋放(圖5g)。總之,上述結果表明[Ca2 +]攝取后形成CPPs會誘導炎癥小體激活IL-1β釋放,且具有濃度依賴性。
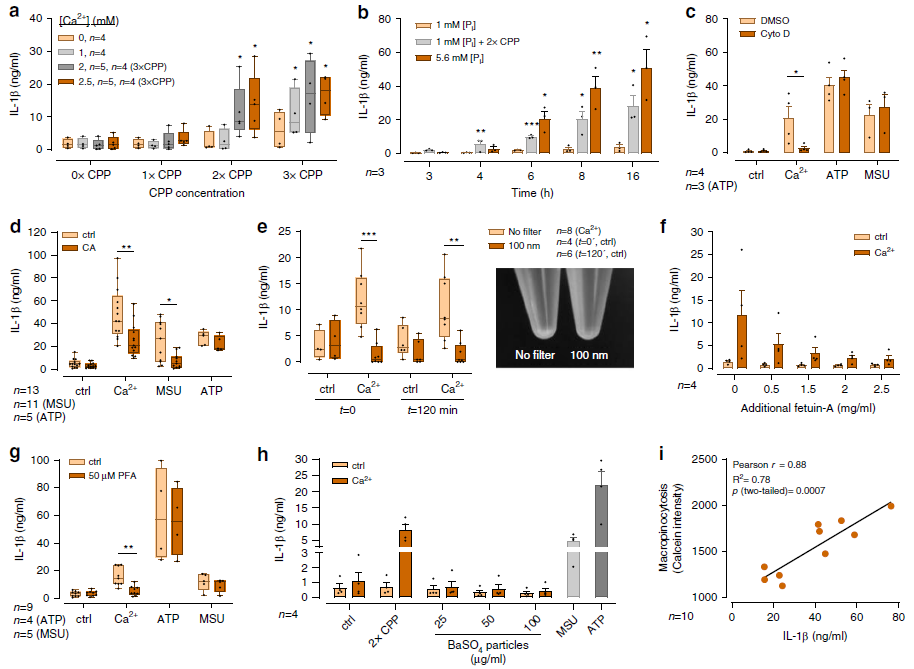
Fig. 5 [Ca2+]ex mediated calciprotein particle uptake mediates inflammasome activation
4、[Ca2+]ex誘導的IL-1β釋放在RA中是增加的
為了進一步證實體內炎癥與對[Ca2 +] ex和CPPs反應之間的聯系,作者進行了體內實驗,結果發現[Ca2+]ex增加的幾次導致RA單核細胞的IL-1β釋放高于健康供體(Fig. 6a),并且無疾病緩解性抗風濕藥(DMARD)的RA患者在用[Ca2 +] ex刺激后產生更高的IL-1β濃度。 除了IL-1β,與健康供體相比,在用[Ca2 +] ex刺激后,RA單核細胞還釋放了更高濃度的IL-1α和IL-18(Fig. 6b,c)。DMARD的RA患者在用[Ca2+] ex刺激后產生更高的IL-1β濃度。 除了IL-1β,與健康供體相比,在用[Ca2+] ex刺激后,RA單核細胞還釋放了更高濃度的IL-1α和IL-18(Fig.b,c)。與來自健康供體的單核巨噬細胞相比,當單核巨噬細胞在體外從RA單核細胞分化時,在[Ca2+]ex刺激后,它們也表現出更高的IL-1鉀離子濃度(Fig. 6e)。較高比例的RA單核細胞內化鈣素染色表明它們增加的傾向是響應[Ca2+]ex,但只對LPS刺激響應(Fig. 6f)。Fig. 6g證實RA中[Ca2+]ex誘導的IL-1β釋放和CPPs攝取導致可能是因為CaSR信號的增加導致的。
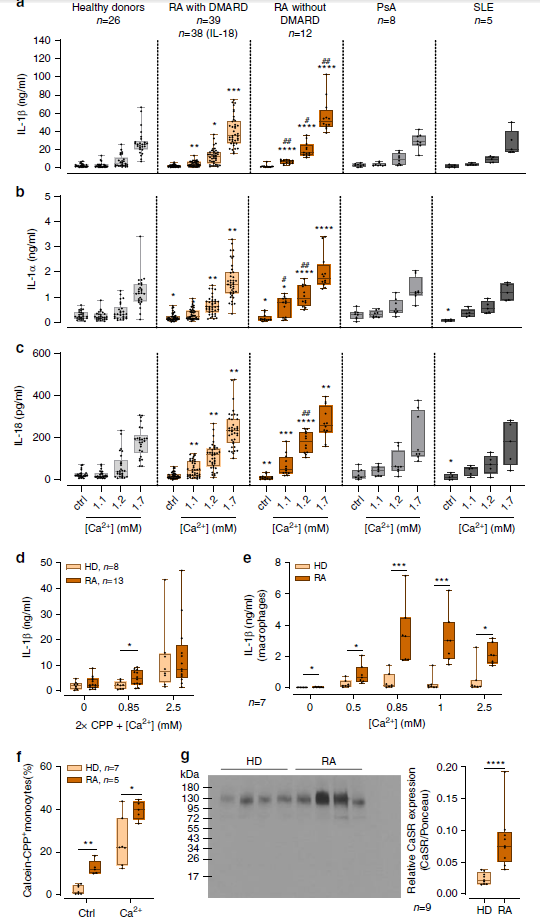
Fig. 6 Increased [Ca2+]ex-induced, CPP-dependent inflammasome activation in RA.
為了研究溶酶體滲漏是否有助于[Ca2+]ex誘導的NLRP3炎性小體活化,將溶酶體用stain啶橙染色,并在存在5.6mM Pi的情況下用2.5mM Ca2 +刺激,或用MSU晶體或溶酶體破壞劑刺激L-亮氨酰-L-亮氨酸甲酯(LLOMe)作為陽性對照。成像流式細胞儀顯示,與陰性對照相比,用[Ca2+]ex刺激后,溶酶體膜的完整性沒有改變(Fig. 7a)。 相比之下,MSU晶體和LLOMe誘導了溶酶體滲漏,由孵育4小時后明亮細節熒光強度顯著降低所表明。RA單核細胞與健康供體之間的溶酶體滲漏比較沒有顯示差異(Fig. 7a)。
通常認為炎癥小體激活和隨后的IL-1β釋放與細胞焦亡或炎性細胞死亡有關,這促使我們研究細胞存活。流式細胞儀(Fig. 7b,c)和標準LDH釋放測定(Fig. 7d)分析的碘化丙啶(PI)/ Hoechst染色均表明,在發生IL-1β釋放的條件下,細胞死亡增加。來自RA患者的單核細胞在單獨用LPS刺激后顯示出增加的細胞存活率,但由[Ca2+]ex或ATP誘導的單細胞死亡率與健康供體無差異(Fig. 7d)。在上述健康的供體中,在升高的[Ca2 +]/Pi條件下細胞死亡率與相應的IL-1β釋放密切相關(Fig. 7e)。細胞死亡部分取決于NLRP3(Fig. 7f)和CaSR(Fig. 7g)的存在。結果表明,抑制CaSR介導的CPP攝取或抑制溶酶體CPP分解可顯著抑制細胞死亡(Fig.7h)。總之,CaSR介導的CPP攝取誘導的炎癥小體激活和隨后的IL-1β釋放與細胞死亡有關。
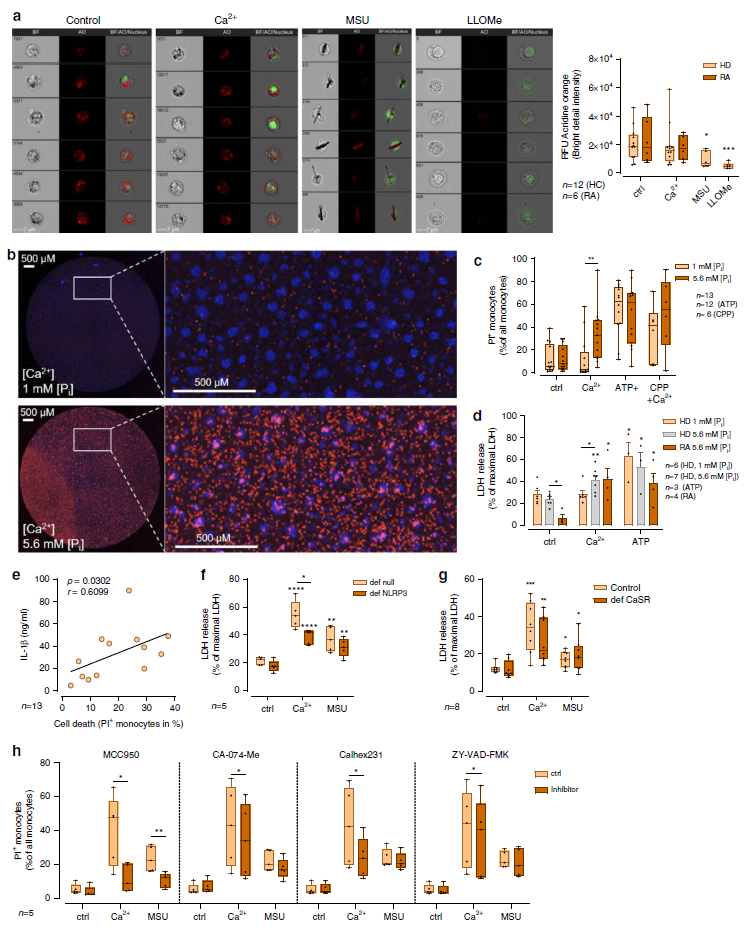
Fig. 7 [Ca2+]ex-induced CPP uptake in monocytes does not induce lysosomal leakage.
5、[Ca2+]ex誘導的IL-1β釋放作用于局部炎癥
接下來,研究了關節炎對[Ca2 +]和[Ca2 +] ex誘導的IL-1β分泌的影響。RA患者的滑液樣本中[Ca2 +]濃度高于骨關節炎或非侵蝕性關節疾病患者的關節積液(Fig.8a)。此外,與骨關節炎樣品相比,在RA患者的滑膜襯里層中發現CaSR表達上調(Fig.8c)。為了研究CaSR在體內關節炎中的作用,將變構CaSR調節劑R568用于DBA / 1J小鼠的膠原蛋白抗體誘發的關節炎(CAIA)模型中。圖8d所示的結果表明,在該小鼠模型中,CaSR信號轉導的正調節加重了關節炎。
為了進一步研究[Ca2 +]在糜爛性關節炎中的作用,首先,從外周血,骨髓和腹膜腔中分離CD11b +單核細胞,以確認糜爛性關節炎與單核細胞IL-1β對[Ca2 +] ex升高的反應之間的聯系。在所有研究的細胞群中,與對照小鼠相比,CIA小鼠的細胞顯示出更強的[Ca2 +] ex誘導的IL-1β反應(Fig.8e)。切片顯示鈣紅染色在骨侵蝕部位和軟骨剝奪的關節表面呈陽性染色(Fig.8f)。因此,通過從股骨中清除骨髓并測量這些骨髓潮紅中的[Ca2 +]來確定CIA中骨骼中Ca2 +的釋放。與對照小鼠相比,關節炎DBA / 1J小鼠的髓內[Ca2 +]顯著增加(Fig.8g),這表明Ca2 +從CIA的骨基質中釋放的增加。 [Ca2 +] ex誘導的IL-1β釋放與骨髓潮紅中確定的[Ca2 +]量(Fig.8h)和CIA評分確定的關節炎嚴重程度(Fig.8i)相關。
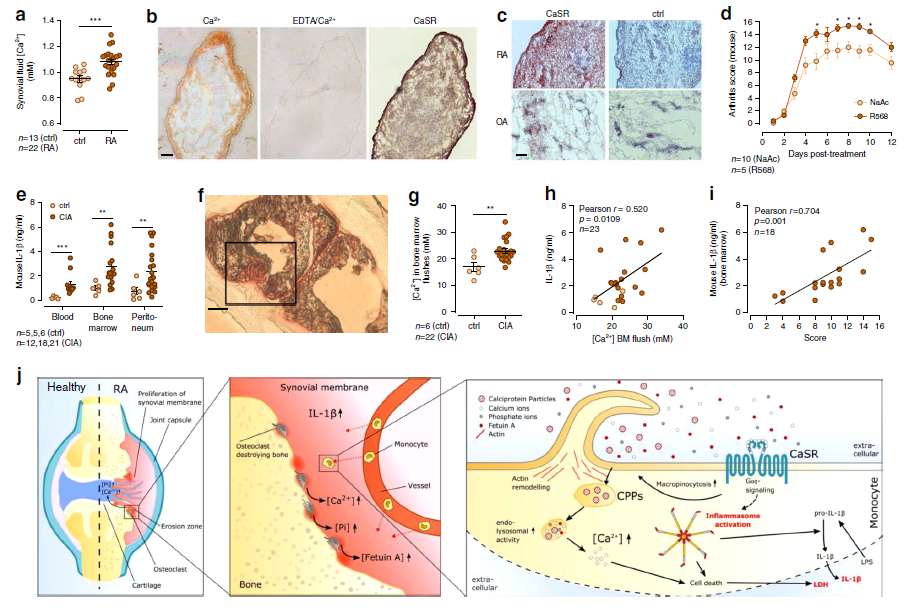
Fig. 8 Bone erosion in arthritis leads to locally increased [Ca2+] and elevated IL-1β secretion.
總之,本文發現單核細胞通過大胞飲作用吞噬CPPs,并且此過程嚴格取決于[Ca2 +] ex的增加觸發的CaSR信號傳導。CPP的巨胞飲作用增強,導致溶酶體活性增加,NLRP3炎性體激活和IL-1β釋放。類風濕關節炎(RA)中的單核細胞響應CaSR信號轉導顯示CPP攝取增加和IL-1β釋放。這些單核細胞和受累關節中局部[Ca2 +]誘導的CaSR表達增加,可能有助于這種增強的炎癥反應。CaSR介導的NLRP3炎性小體活化不僅在RA中而且在其他炎性疾病中也可引起炎性關節炎和全身性炎癥。抑制CaSR介導的CPP攝取可能是治療RA的治療方法。
參考文獻:
J?ger Elisabeth., Murthy Supriya., Schmidt Caroline., Hahn Magdalena., Strobel Sarah., Peters Anna., St?ubert Claudia., Sungur Pelin., Venus Tom., Geisler Mandy., Radusheva Veselina., Raps Stefanie., Rothe Kathrin., Scholz Roger., Jung Sebastian., Wagner Sylke., Pierer Matthias., Seifert Olga., Chang Wenhan., Estrela-Lopis Irina., Raulien Nora., Krohn Knut., Str?ter Norbert., Hoeppener Stephanie., Sch?neberg Torsten., Rossol Manuela., Wagner Ulf.(2020). Calcium-sensing receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat Commun, 11(1), 4243. doi:10.1038/s41467-020-17749-6