






肝星狀細胞自噬抑制EV釋放改善肝纖維化
研究背景:
自噬是真核生物的一種降解過程,稱為自噬體的雙膜小泡與溶酶體融合以降解胞漿蛋白和細胞器。自噬有助于肝內環境的穩定,其調節失調與一些慢性肝臟疾病有關。在肝細胞中,自噬已被證明具有保護作用,而在肝星狀細胞(HSCs)中,自噬被提出通過脂噬(一種選擇性的脂滴降解)誘導其激活。然而,自噬抑制劑雷帕霉素靶點(mTOR)促進星狀細胞的活化和肝纖維化。因此,破譯自噬參與的纖維化信號放大微調機制將提高我們對肝纖維化的理解。
在肝臟中,受損肝細胞和竇狀內皮細胞(LSECs)產生的細胞外囊泡(EVs)可誘導HSC的活化和遷移。有趣的是,肝損傷后EV釋放增加,而自噬抑制或溶酶體降解抑制與EV釋放增加有關。然而,自噬如何調節前纖維化的HSC來源的EV的生物發生,目前仍缺乏了解。
在本研究中,作者證明了HSCs通過mTOR抑制自噬和激活RHOR相關蛋白激酶1 (ROCK1)信號來釋放EV。此外,這種EV釋放機制參與了肝纖維化進程。 總之,HSC自噬通過減少纖維化HSC來源的EV釋放來減輕肝纖維化。本文于2020年5月7日發表在Journal of Hepatology(IF:18.946)雜志上。
結 果:
1、PDGF抑制自噬,增加EV釋放
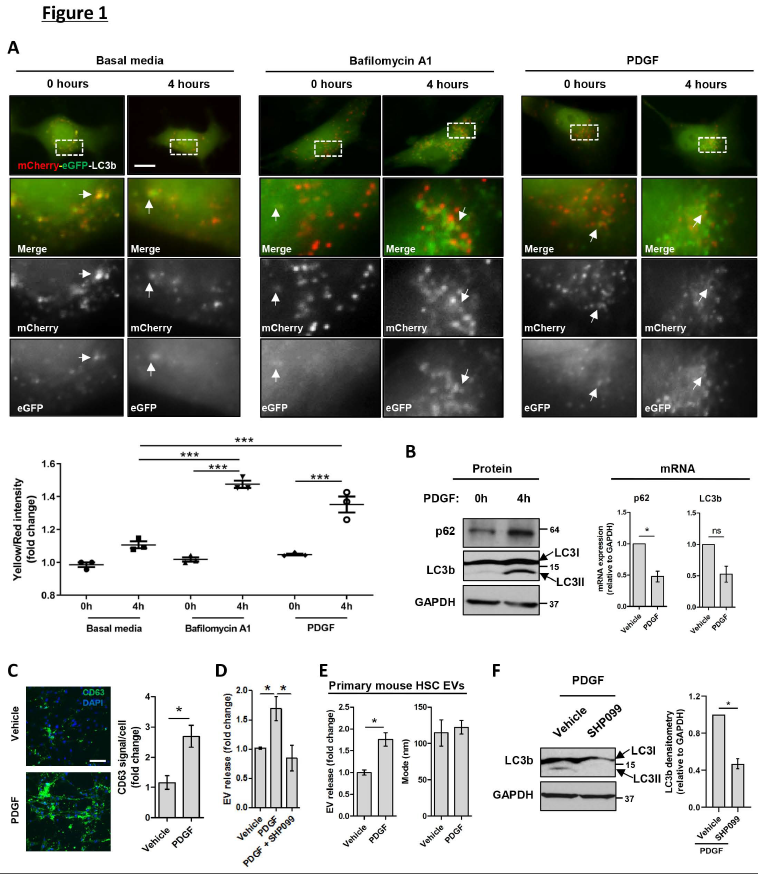
首先驗證假設,在HSCs中,PDGF刺激模型抑制自噬以增加EV釋放。用mCherry-eGFP-LC3b質粒轉染LX2細胞(人肝星狀細胞系),用自噬降解抑制劑bafilomycin A1或PDGF處理LX2細胞。正如預測的,與載體(圖1A左)相比,bafilomycin A1處理增加了eGFP+-mCherry+自噬體池(黃色)(圖1A中),導致黃/紅比例增加(圖1A圖),提示自噬抑制。與bafilomycin A1相似,與基礎培養基比較,PDGF在處理4h時提高了黃/紅比例(圖1A右),首次證明PDGF抑制自噬降解。
與這些結果一致,PDGF處理導致人肝星狀細胞的自噬體LC3b和sequestosome-1 (p62) 蛋白質含量增加,但PDGF并沒有增加LC3b或p62 mRNA水平(圖1B,右),這表明蛋白質水平的增加是由于自噬降解的減少,而不是由于p62和LC3b的重新轉錄表達。PDGF介導的自噬抑制與原代HSC細胞MVB池的CD63+增加(圖1C),以及人和小鼠HSC細胞來源的EV釋放的增加相關(圖1 d- e)。PDGF的關鍵下游信號分子之一是SHP2,通過競爭性抑制劑SHP099抑制SHP2,消除PDGF介導的EV釋放(圖1D)。SHP099還降低了LC3b蛋白水平(圖1F),表明SHP2抑制促進了自噬降解。這些結果支持了HSCs中PDGF信號抑制自噬并增加EV釋放的觀點,提示自噬可能抑制EV釋放。
2、EV釋放受自噬激活因子REDD1調控
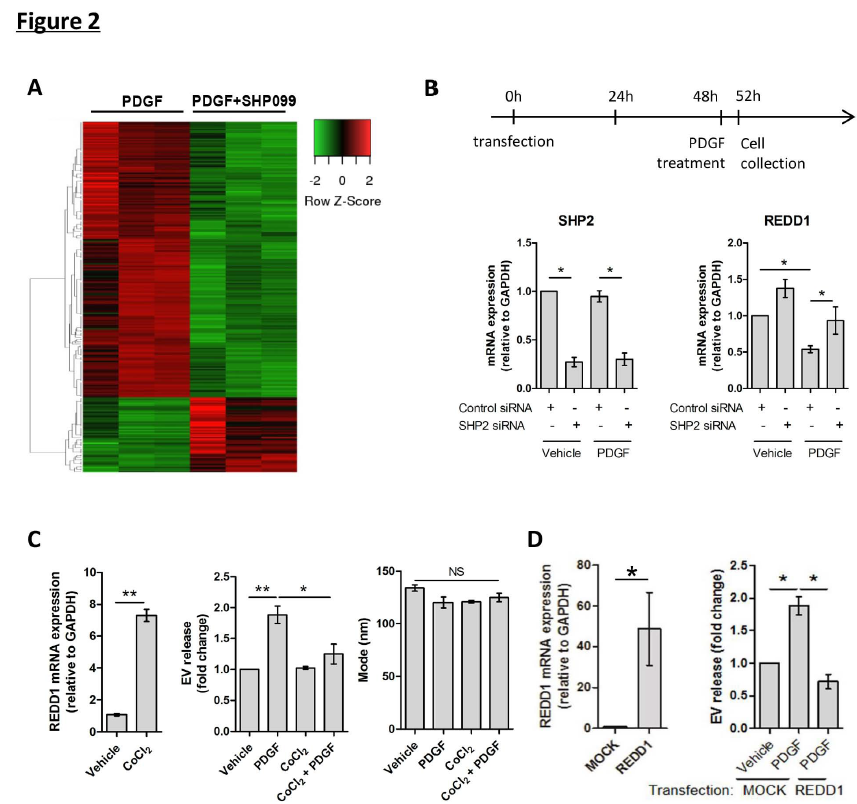
使用PDGF/SHP2模型進一步研究自噬如何抑制纖維性EV釋放。為此,用PDGF+/-SHP099處理人原發性HSCs,然后全轉錄組RNA測序檢測 (圖2)。近25%的基因被SHP099上調(圖2A)。結果得到REDD1,REDD1是mTOR的內源性抑制劑,是SHP2的首要靶點之一。在對照和PDGF處理和siRNA敲除SHP2均導致REDD1上調(圖2B)。 這些結果表明,PDGF和SHP2負調控mTOR抑制劑和自噬激活劑REDD1的表達,提示REDD1在EV釋放中的作用。
接下來,通過刺激REDD1的表達來了解REDD1在肝纖維化和HSC衍生的EV釋放中的作用。使用REDD1刺激物 CoCl2(圖2C,左)。在EV或PDGF存在下,用CoCl2處理原代人HSC。REDD1表達減弱了PDGF介導的EV釋放(圖2C,中間),而對EV大小沒有影響(圖2C,右)。接下來,我們在LX2細胞中過表達REDD1(圖2D,左)。REDD1的過表達顯著降低了HSC衍生的EV釋放(圖2D,右)。 總之,這些結果表明,REDD1是一種自噬激活劑和mTOR抑制劑,是HSC來源的纖維化EV釋放的負調節劑。
3、mTOR信號通過抑制自噬和激活ROCK1來增加HSC誘導的EV釋放
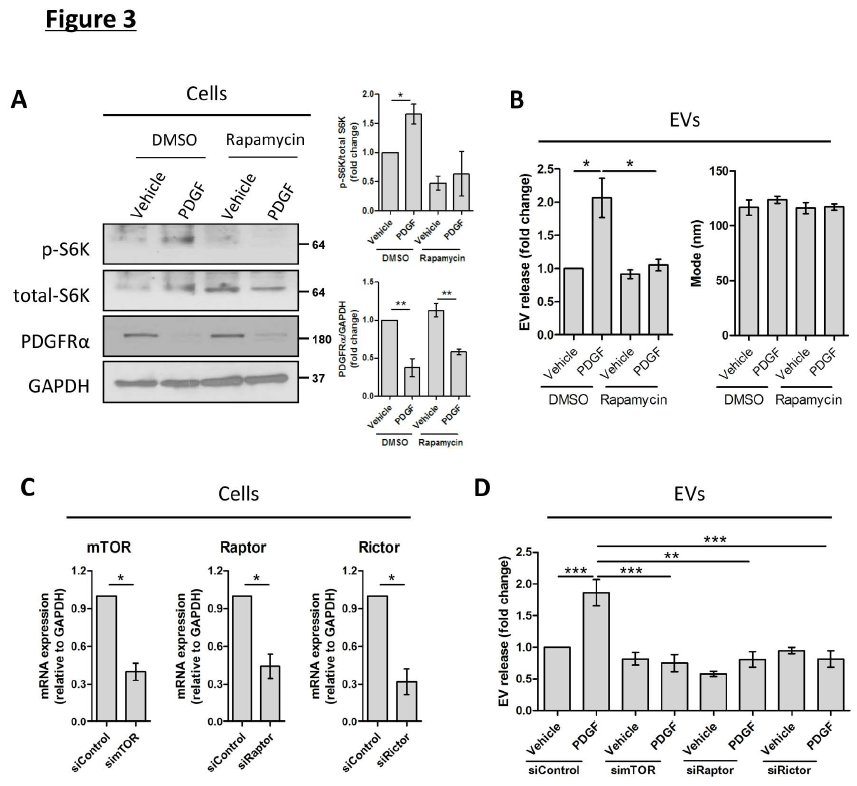
為了進一步了解自噬如何影響纖維化EV釋放,研究了自噬抑制劑mTOR在HSC衍生的EV釋放中的作用。結果表明,雷帕霉素對mTOR的抑制作用減弱了PDGF介導的EV釋放,而對EV的大小沒有任何影響(圖3B)。 然后,在人HSC上進行了mTOR結合蛋白沉默,mTOR的調節相關蛋白(Raptor)和mTOR雷帕霉素不敏感相關蛋白(Rictor)沉默(圖3C)。 與雷帕霉素結果一致,siRNA引起的mTOR敲除消除了PDGF介導的EV釋放(圖3D)。 Raptor或Rictor siRNA介導的敲低也獲得了類似的效果(圖3D),證實mTOR信號傳導促進EV釋放。
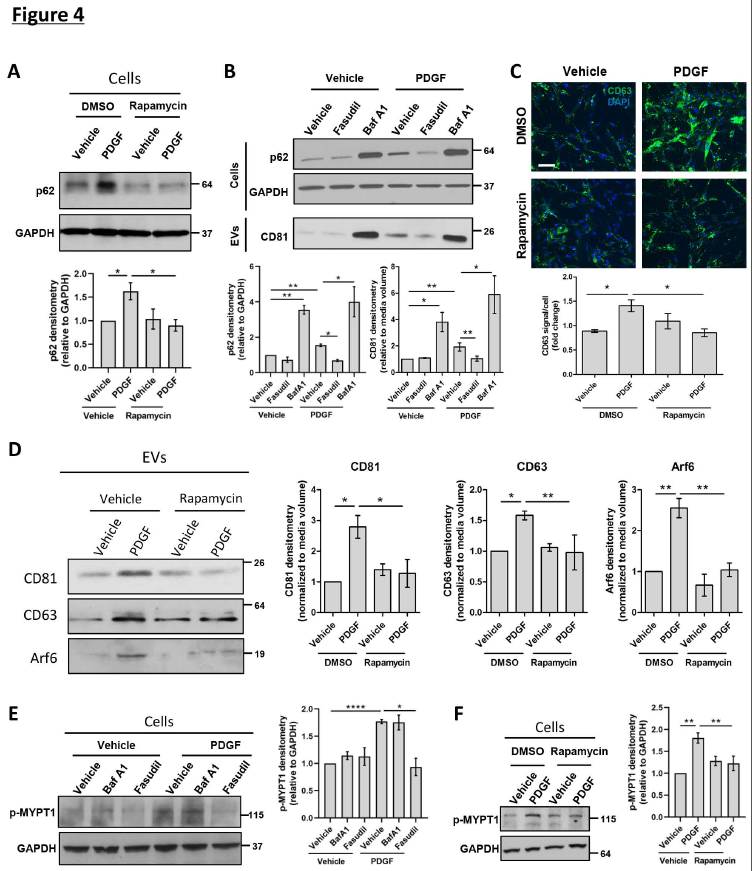
接下來,研究了原代人HSC中mTOR依賴的EV釋放的起源。如圖PDGF增加了p62蛋白水平,該水平被雷帕霉素減弱(圖4A),表明PDGF通過mTOR抑制自噬。此外,通過bafilomycin A1(圖4B)造成的自噬抑制作用顯著增加了EV級分中的外泌體標記物CD81以及微泡標記物Alf6的表達。根據這些結果,作者研究了mTOR信號在原代人HSC中MVB的作用,其中CD63是MVB和外泌體標記。用PDGF處理細胞可增加MVB,雷帕霉素可將其消除,如免疫熒光法所證實(圖4C)。與此相一致,雷帕霉素降低了PDGF介導的外泌體釋放,如EV中CD81和CD63蛋白水平的降低得以證明(圖4D)。這些結果表明,mTOR信號傳導誘導外來體釋放。
與外泌體相反,微泡釋放取決于ROCK1的活性。因此,接下來研究了mTOR信號傳導對ROCK1活性和微泡釋放的影響。PDGF處理原發性人HSC細胞激活了ROCK1信號,表現為ROCK1下游信號分子,肌球蛋白磷酸酶靶標亞基1(MYPT1)的磷酸化水平升高(圖4E)。通過用ROCK1抑制劑fasudil處理HSC可以消除這種情況(圖4E)。隨后,fasudil對ROCK1的抑制作用顯著降低了PDGF介導的EV釋放,如微泡標志物Arf6和外泌體標志物CD81表達下調,表明外泌體和微泡釋放機制之間存在相互影響。PDGF介導的ROCK1活性也被雷帕霉素抑制,如MYPT1磷酸化減弱(圖4F)表明PDGF通過mTOR誘導ROCK1活化。此外,雷帕霉素降低了PDGF介導的微泡釋放,這通過降低EV餾分中的Arf6蛋白水平來證明(圖4D)。這些數據表明,mTOR信號傳導誘導微泡釋放。
總之,mTOR信號通過抑制MVB的自噬降解誘導外泌體釋放,并通過激活ROCK1信號傳導來誘導微泡釋放,提供了對PDGF介導的纖維化EV釋放的機制的見解。
4、mTOR介導的EV誘導HSC細胞的體外遷移
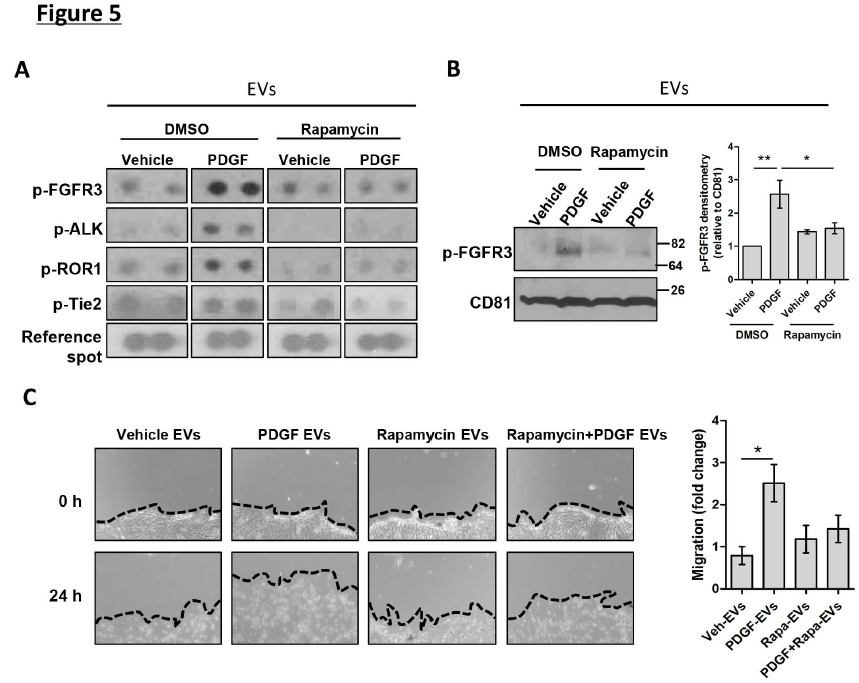
受體酪氨酸激酶(RTK)信號在纖維化中占據關鍵地位,因此我們利用磷酸化RTK蛋白陣列評估了EV含量。RTK陣列結果顯示,PDGF處理后的EV中存在幾種磷酸化的RTK,例如成纖維細胞生長因子受體3(FGFR3),內膜內皮細胞激酶(Tie2),間變性淋巴瘤激酶(ALK)和受體酪氨酸激酶-孤兒受體1(ROR1)(圖5A)。受PDGF上調,被雷帕霉素降低最顯著的是磷酸FGFR3(圖5A),它參與成纖維細胞遷移。這些數據表明,mTOR信號不僅促進了EV的釋放,而且還通過激活RTK介導EV的富集。由于以前已確定RTK與細胞遷移有關,所以接下來驗證這些富含RTK的EV對HSC遷移的影響。與衍生自對照細胞的EV相比,衍生自PDGF處理的EV顯著增加了受體HSC的遷移(圖5C)。總之,這些結果表明,mTOR促進了轉移前的RTK富集的EV的釋放。
5、體內刪除HSC特異性SHP2肝纖維化下降
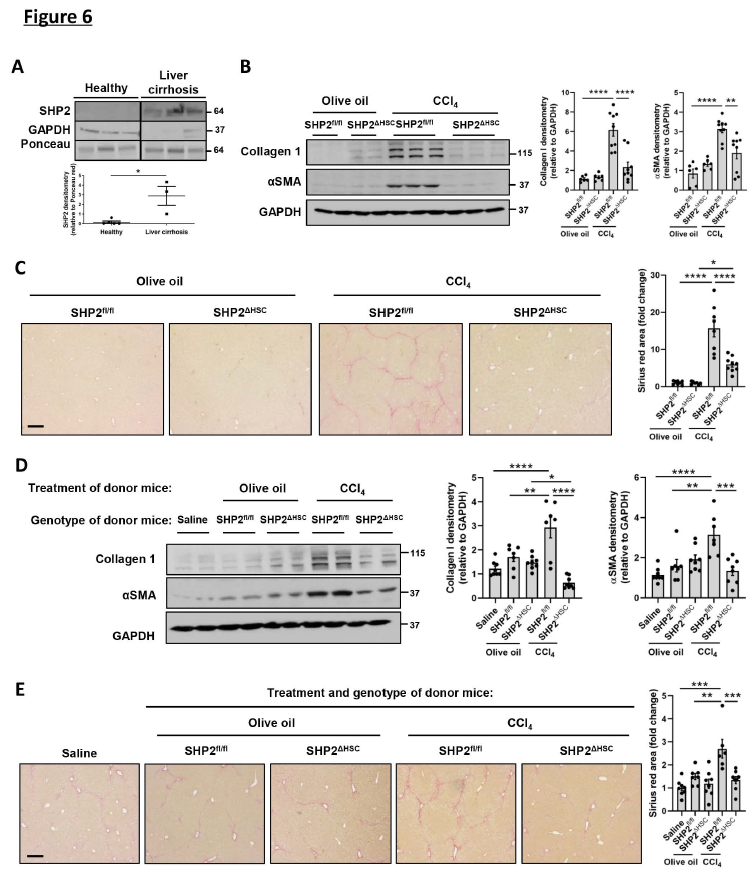
由于SHP2是mTOR依賴性EV釋放的上游調節劑,在全肝裂解物中,與健康個體相比,肝硬化患者的SHP2升高(圖6A)。因此,驗證HSC特異性SHP2缺失對肝纖維化建立的影響,由 CCl4 給藥6周誘導肝纖維化。然后收集肝臟,并用天狼星紅染色和WB進行分析。如預期的那樣,慢性CCl4處理后同窩對照小鼠(SHP2f1 / f1)的肝膠原蛋白I,αSMA蛋白水平和天狼星紅染色增加(圖6B-C)。然而,如膠原蛋白I,αSMA和天狼星紅染色的蛋白質水平所表明的,當在HSC(SHP2ΔHSC小鼠)中選擇性刪除SHP2基因時,肝纖維化顯著減少。總之,HSCs中SHP2的選擇性缺失顯著減弱了肝纖維化。
6、HSC特異性 SHP2敲除的老鼠EV減少了WT老鼠中 CCl4介導的肝纖維化
前文證明,由PDGF處理的HSC衍生的EV具有促纖維化作用,而SHP2對肝纖維化至關重要(圖6B-C)。因此,接下來研究HSC特異性SHP2如何影響循環EV的纖維化潛力,以及這些EV如何在體內影響肝纖維化。與生理鹽水對照相比,橄欖油處理的SHP2fl / fl或SHP2ΔHSC供體小鼠的EV不會影響肝纖維化。 但是,如WB和Sirius red(圖6D-E)所示,來自經CCl4處理的SHP2fl / fl對照小鼠的EV顯著增加了CCl4誘導的肝纖維化(圖6D-E),表明這些EV具有成纖維性。 此外,從CCl4處理的SHP2ΔHSC小鼠中分離出的EV顯著減少了CCl4誘導的肝纖維化(圖6D-E),這表明在HSC中選擇性刪除SHP2會降低EV的纖維化潛力。 總之,HSC中SHP2的選擇性缺失顯著減弱了循環EV的纖維化特性。
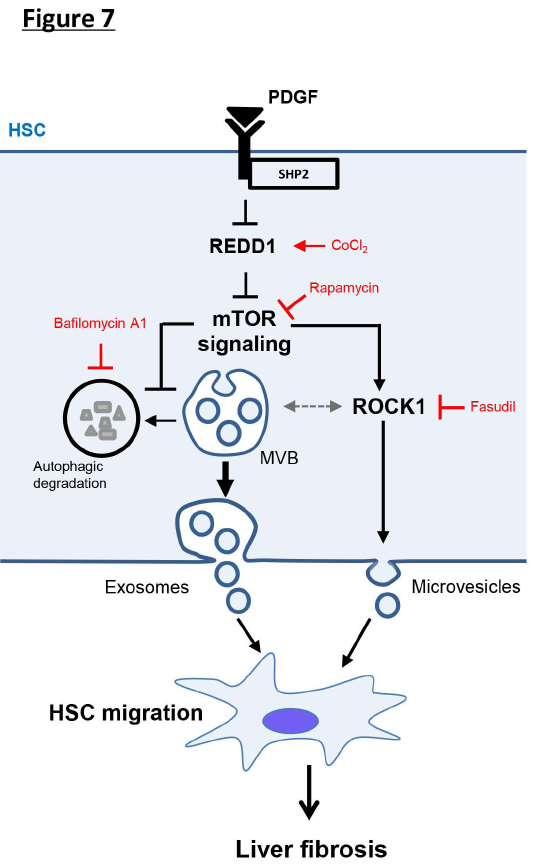
總之,HSC中的SHP2通過抑制自噬,REDD1和激活mTOR途徑來增強纖維化電動汽車的釋放,從而發揮其促纖維化作用。 SHP2-mTOR信號的抑制可能作為治療肝纖維化的新靶標。