






肥胖病研究新火花:m6A甲基化可調控自噬和脂肪形成
肥胖,一類由脂肪細胞的體積和數量增加所引起的脂肪組織的異常或過度積累的疾病,已然成為人們關注的焦點,提高對于脂肪生成分子機制的理解具有重要的科學意義。研究人員已提出許多調節脂肪細胞分化的機制,包括細胞外信號,轉錄級聯和表觀基因修飾等。近日,這篇文章提出了肥胖研究的新奇思路,甲基化和自噬之間能碰撞出什么火花呢,我們來一睹為快吧。

文章思路:

1.FTO促進自噬并進一步增強脂肪形成
探討FTO在自噬中的作用:3T3-L1細胞中FTO敲低顯著降低LC3-II:I比值和增加SQSTM1水平,表明沒有穩態自噬體形成。相反,過度表達HA標記的FTO顯著提高LC3-II:I比值和減弱SQSTM1表達,表明FTO與自噬之間存在正相關關系。FTO的沉默減少自噬體的數量, 減輕自噬的激活。為確定自噬是否受FTO過表達影響, 3-MA或巴弗洛霉素A1處理實驗進一步證實FTO促進自噬的激活。
檢測FTO是否通過自噬影響脂肪形成: 3-MA和Baf A1處理可以逆轉FTO過表達細胞中增強的自噬、脂肪生成和甘油三酯的積累。脂肪細胞標記基因,包括Pparg、Fabp4和Cebpa在FTO過表達細胞中顯著升高,3-MA或Baf A1處理可以下調到正常水平。這些結果表明FTO通過促進自噬增強脂肪細胞分化。
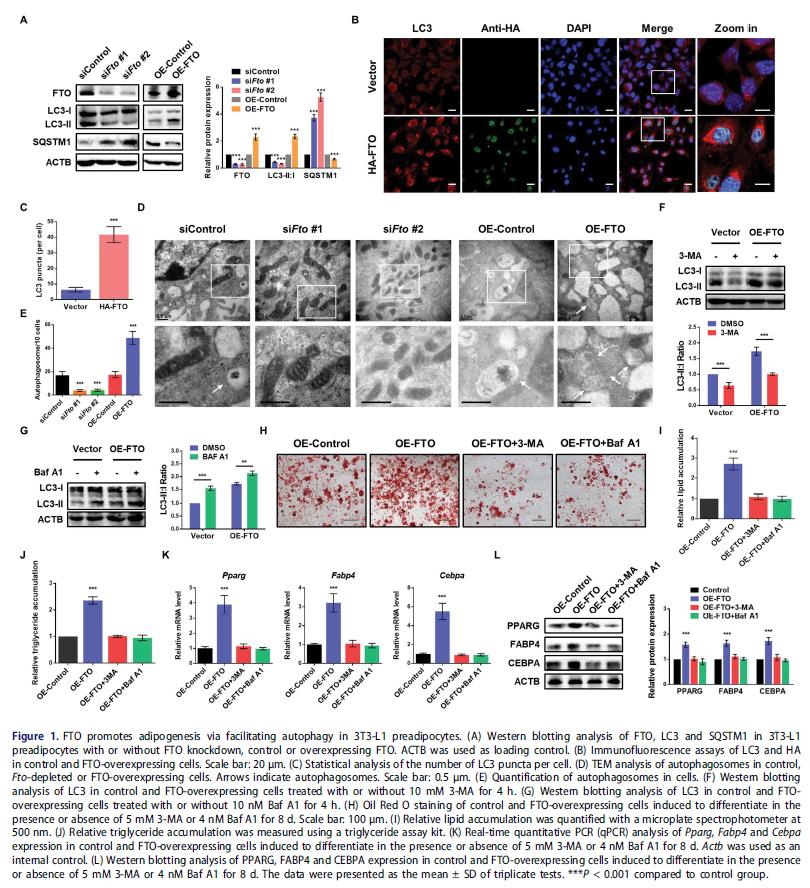
2. 敲低FTO可降低ATG5和ATG5的表達,但沒有降低ULK1的表達
鑒定FTO在自噬中的潛在靶基因:檢測FTO敲低后自噬相關基因的表達,Atg5和Atg7水平均減弱,而ULK1等自噬相關蛋白,如ATG12和ATG16L1,沒有顯著改變。檢查脂肪細胞分化過程中ATG5和ATG7的表達譜,發現它們都在早期有所增加,之后都減少,這與FTO的表達模式相似。這些表明FTO的靶基因可能是Atg5和Atg7,而不是Ulk1。
檢測ATG5和ATG7對自噬和脂肪生成的影響:3T3-L1脂肪前體細胞敲低Atg5和Atg7, LC3II:I比值降低, FTO蛋白表達沒變化。FTO,ATG5和ATG7獨自敲低都會抑制脂肪生成和甘油三酯積累,脂肪細胞標記基因水平下調。
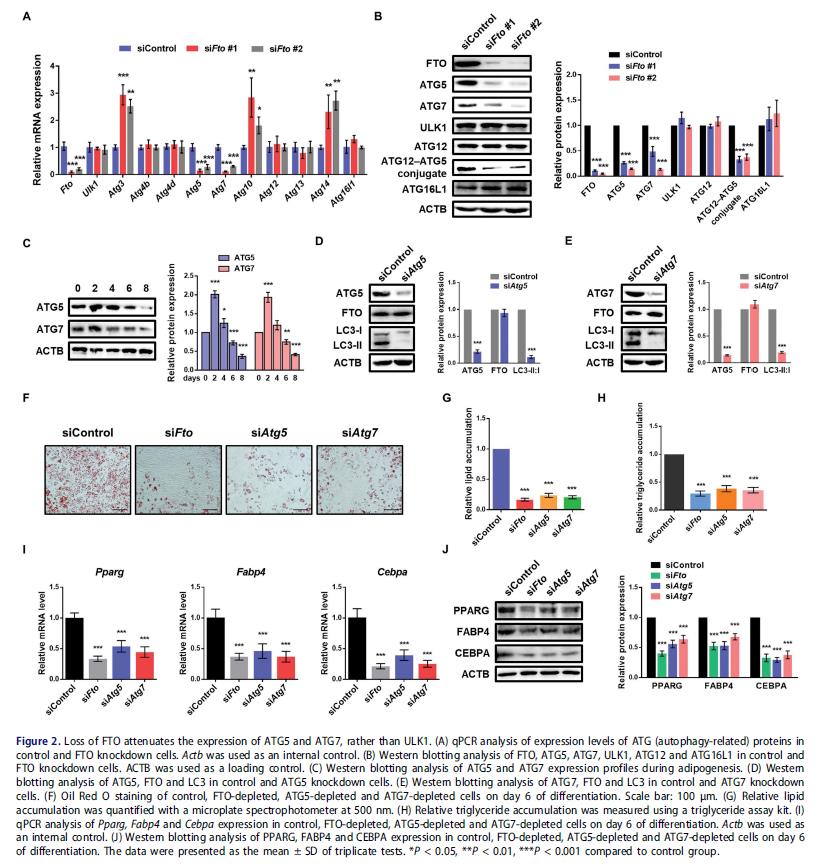
3.FTO靶向 ATG5和ATG7并影響自噬和脂肪形成
FTO過表達增加Atg5和Atg7的mRNA水平,沉默ATG5可逆轉FTO過表達3T3-L1細胞中上調的LC3-II:I比例, ATG7效果一樣,表明FTO通過調節ATG5和ATG7的表達影響自噬。前人研究發現ATG5和ATG7敲低抑制自噬并減少CEBPB水平,抑制PPARG和CEBPA表達損害脂肪形成分化,提示FTO通過Atg5和Atg7-Cebpb信號調節脂肪生成。FTO強表達促進CEBPB蛋白水平,而ATG5和ATG7的敲低逆轉CEBPB的表達(圖3D)。ATG5和ATG7的敲低恢復了FTO過表達3T3-L1細胞的脂肪生成 和甘油三酯積累, FTO過表達細胞中Pparg,Fabp4和Cebpa的mRNA和蛋白水平也被逆轉。FTO通過調節Atg5和Atg7-Cebpb信號軸促進脂肪生成。
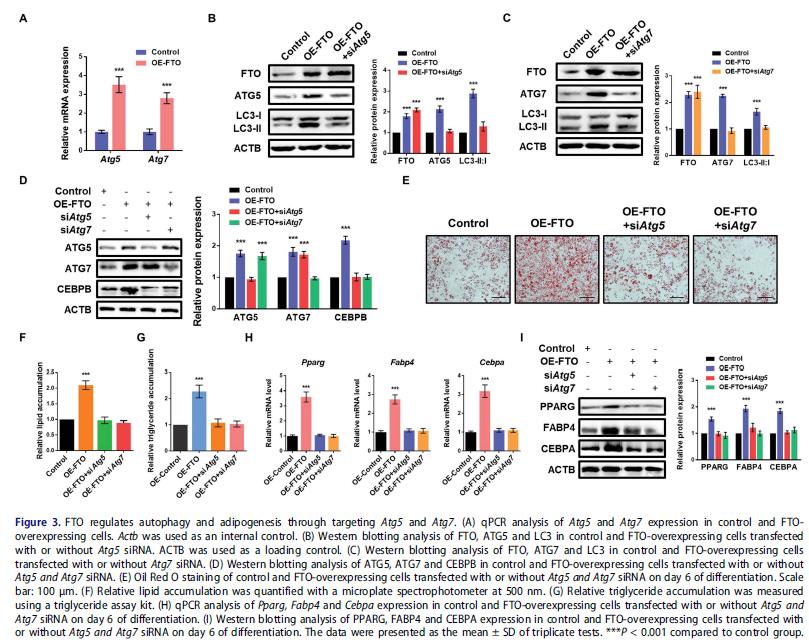
4.FTO以 m6A依賴方式調節ATG5和ATG7的表達
構建野生型(FTO-WT)和突變的FTOR96Q(FTO-MUT)質粒,來確定FTO的去甲基酶活性是否必須。LC-MS / MS確認FTO-WT或FTO-MUT的異位表達對細胞m6A水平的影響。異位表達FTO-WT,但不是FTO-MUT,顯著增加LC3-II:I表達,LC3斑點顯著增強,自噬體的數量提高。這些結果證明FTO去甲基化的活性是前脂肪細胞中自噬所必需的。
調查FTO通過RNA去甲基化影響ATG5和ATG7表達,異位表達FTO-WT增加ATG5和ATG7的蛋白質和mRNA水平,而ULK1蛋白豐度沒有變化。在Atg5和Atg7的3'UTR處發現m6A修飾。發現FTO的敲除增加3T3-L1細胞的m6A水平。基因特異性甲基化RNA免疫沉淀-qPCR(MeRIP-qPCR)表明FTO敲低顯著增加Atg5和Atg7 mRNA轉錄物的m6A水平,但不對Ulk1有影響。評估靶mRNA進行m6A修飾對于FTO介導的基因調控是否必要,3T3-L1細胞中進行雙熒光素酶報告和誘變實驗。強表達FTO-WT,但不是FTO-MUT,大大促進含有Atg5和Atg7的3'UTR片段的單個報告結構的熒光素酶活性,當m6A位置發生突變時,這種增加被消除,證明FTO通過m6A依賴機制調節ATG5和ATG7的表達。油紅O染色分析顯示FTO-WT促進脂肪細胞分化和甘油三酯積累,驗證去甲基化脂肪形成需要FTO的活性。這些結果說明FTO靶向Atg5和Atg7轉錄物并以m6A依賴的方式介導他們的表達,進一步調節自噬和脂肪生成。
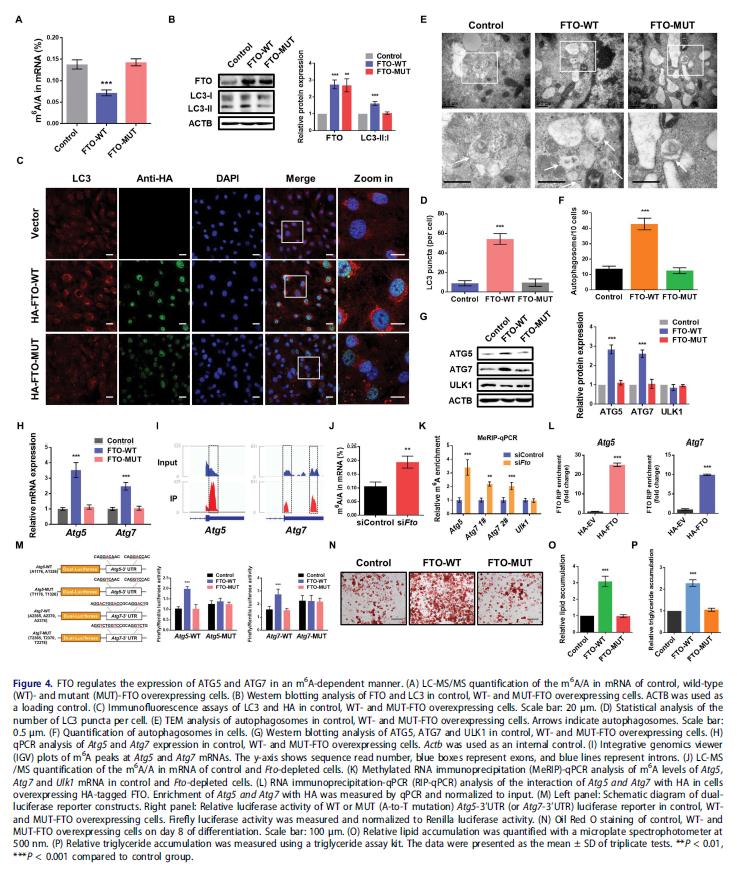
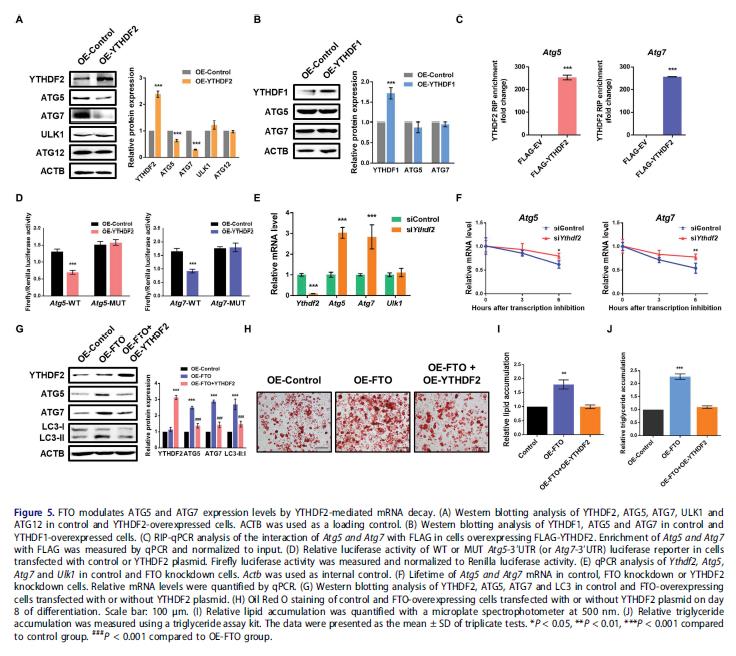
5.YTHDF2通過m6A依賴機制介導ATG5和ATG7的mRNA穩定性和表達
m6A甲基化影響mRNA穩定性和翻譯,YTHDF2選擇性識別m6A修飾的mRNA并使其不穩定,促進目標mRNA的翻譯。進一步解釋FTO靶基因的m6A甲基化和蛋白質豐度之間的負相關,首先研究ATG5和ATG7表達是否受YTHDF2或YTHDF1的影響,YTHDF2過表達顯著降低ATG5和ATG7蛋白水平,而ULK1和ATG12無變化,強表達YTHDF1不影響ATG5和ATG7的表達。
檢測在Atg5和Atg7 mRNAs的m6A修飾對YTHDF2介導的基因調控是是否必需的。YTHDF2異位顯著下調野生型Atg5和Atg7片段的熒光素酶活性,這種減少完全被m6A共識位點的突變消除。檢測YTHDF2是否介導mRNA衰變從而控制Atg5和Atg7的表達,在3T3-L1細胞中進行功能缺失研究。敲低YTHDF2后 Atg5和Atg7的mRNA水平提高, mRNA穩定性分析顯示YTHDF2缺失延長了Atg5和Atg7 mRNA轉錄物的半衰期。過表達FTO的3T3-L1細胞中ATG5和ATG7蛋白質水平的增加能部分被強表達YTHDF2逆轉,LC3-II:I也部分逆轉。異位表達YTHDF2可逆轉過表達FTO引起的促脂肪生成和甘油三酯積累。這些數據表明FTO調節ATG5和ATG7的表達,通過m6A依賴性和YTHDF2介導的方式。
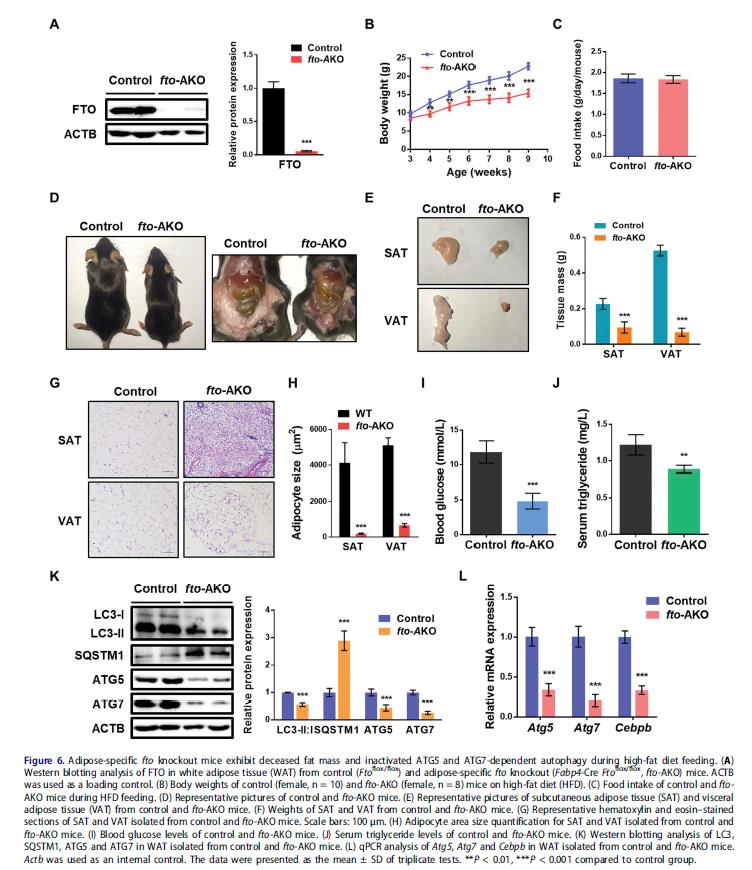
6.脂肪選擇性缺失FTO可減少白色脂肪量并抑制ATG5和ATG7依賴性自噬
研究FTO的體內作用,建立脂肪選擇性FTO敲除小鼠(fto-AKO)模型,fto-AKO小鼠的脂肪組織(WAT)FTO表達缺失,其他組織包括肌肉,肝臟,大腦,巨噬細胞或血管內皮細胞并沒有。相對于對照鼠,食物或高脂飲食(HFD)誘導fto-AKO小鼠的體重增加變弱,且更瘦,而兩組之間食物攝入沒有顯著差異。fto-AKO小鼠腹股溝脂肪和性腺脂肪的數量顯著低于同窩對照鼠。SAT和VAT的haematine和伊紅染色顯示HFD對照小鼠的脂肪細胞表現為一個大脂滴占據整個細胞的典型結構。相反,FTO缺乏導致多腔較小脂滴的脂肪細胞。喂食HFD的fto-AKO小鼠的血糖水平和血清甘油三酯水平的顯著低于對照鼠。脂肪選擇性缺失Fto減弱LC3-II:I比例和SQSTM1蛋白豐度升高,ATG5和ATG7的基因表達減弱,表明在WAT中自噬抑制。此外, fto-AKO小鼠WAT中Cebpb的mRNA水平下調,暗示fto敲除可能損害脂肪組織發育,通過抑制ATG5和ATG7-CEBPB信號。這些表明脂肪細胞選擇性fto敲除可能會減少WAT量和損害ATG5和ATG7依賴性自噬。
總結:
FTO在促進自噬和脂肪生成中起作用,主要通過m6A-YTHDF2依賴機制。這些發現在預防和治療肥胖的研究進程中迸發出新的火花新的思路。