






高2,6-唾液酰化促進CD4+ t細胞活化并誘導潰瘍性結腸炎的發生
2,6-唾液酰化,由2,6-唾液酰轉移酶(ST6GAL1)催化,在免疫應答中起關鍵作用。然而,ST6GAL1在潰瘍性結腸炎(UC)發病機制中的作用尚不清楚。ST6GAL1 mRNA在UC組織中較相應的鄰近正常組織高表達,并且在UC患者結腸組織中2,6-唾液酰化顯著升高。ST6GAL1和促炎細胞因子如白細胞介素(IL)-2、IL-6、IL-17和干擾素- γ的表達也增加。UC患者CD4+ T細胞數量增加。ST6GAL1基因敲除(ST6GAL1?/-)大鼠是通過CRISPR建立的。缺乏ST6GAL1可降低UC模型大鼠的促炎細胞因子水平,減輕結腸炎癥狀。2,6-唾液化的消融抑制TCR向脂質筏的轉運并抑制CD4+ t細胞的活化。TCR信號的衰減可下調ST6GAL1-/- CD4+ t細胞中NF-B的表達。此外,NF-B可以結合ST6GAL1啟動子來增加其轉錄。ST6GAL1的消融術可下調NF-B的表達,減少促炎細胞因子的產生,緩解UC的發病機制,是UC臨床治療的潛在新靶點。該文于2023年9月發布于《Advanced Science》, IF=15.1。
技術路線:

主要研究結果:
1、2,6-唾液酰化在UC中高表達
不同種類的n -聚糖由幾種糖苷酶和糖基轉移酶催化。最重要的“封頂”反應包括通過STs將唾液酸添加到分支上。為了探討唾液酰化與UC發生的相關性,作者使用Gene expression Omnibus (GEO)數據庫生物信息學分析分析了UC患者與健康對照(HCs)中ST家族基因的表達(圖1B)。在GSE179285數據集中,與HCs相比,UC患者中有17044個基因發生改變,其中包括8396個上調基因和8348個下調基因(圖1C)。在UC患者中,ST6GAL1、ST3GAL1、ST3GAL2、ST3GAL3、ST3GAL4、ST3GAL5、ST6GALNAC4、ST6GALNAC5、ST6GALNAC6、ST8SIA1、ST8SIA4的表達水平顯著升高(圖1D)。使用GEO2R分析軟件對樣本間的數據進行歸一化方差分析(圖1E)。在UC或結腸癌患者中觀察到異常的唾液化。作者從UC和HCs患者的結腸組織中分離RNA,比較STs的mRNA水平。與HCs相比,UC患者中ST6GAL1 mRNA表達選擇性過表達(圖1F)。ST6GAL1催化唾液酸從CMP-Neu5Ac供體轉移到半乳糖末端殘基,形成2,6-連鎖(2,6-唾液酰化)(圖1A)。作者用特異性識別2,6-唾液酸結構的黑Sambucus nigra凝集素(SNA)凝集素印跡法檢測了2,6-唾液酸原位化水平。2,6-唾液酰化主要在UC患者的結腸組織中升高(圖1G)。
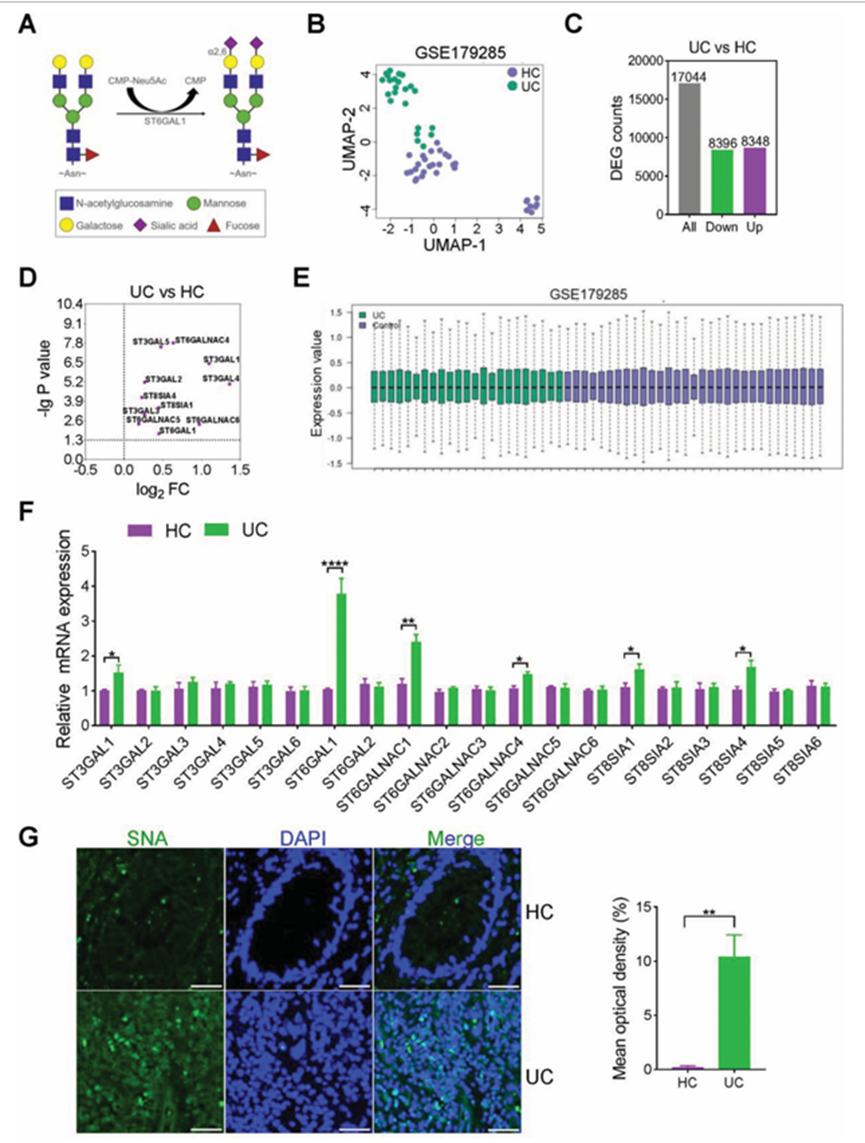
圖1、2,6-唾液酰化在UC結腸組織中顯著升高
2、ST6GAL1消融術緩解大鼠UC癥狀
鑒于2,6-唾液酰化在UC患者中顯著升高,作者研究了ST6GAL1介導的2,6-唾液酰化如何調節UC的發病機制。作者使用CRISPR-Cas9技術改造了ST6GAL1+/?大鼠(圖2A)。通過雜合ST6GAL1+/?大鼠雜交獲得純合野生型(ST6GAL1+/+)和敲除型(ST6GAL1?/?)大鼠(圖2B)。當ST6GAL1+/?大鼠雜交時,ST6GAL1+/+、ST6GAL1+/-和ST6GAL1?/-大鼠的比例≈1:2:1,符合孟德爾遺傳(圖2C)。ST6GAL1產物是2,6-唾液化的n -聚糖,通過SNA印跡證實,它在ST6GAL1 +/+血清中普遍表達,但在ST6GAL1?/?血清中不存在(圖2D)。在凝集素印跡分析中,Aleuria aurantia lectin (AAL)、Maackia amurensis lectin (MAL)和concanavalin A lectin (ConA)的差異無統計學意義。ST6GAL1基因的消融不影響大鼠的體重(圖2E)。為了評估ST6GAL1在UC發生中的作用,作者用5%葡聚糖硫酸鈉(DSS)給ST6GAL1 +/+和ST6GAL1?/?大鼠建立了UC模型(圖2F)。首先,作者用免疫熒光法檢測了大鼠體內2,6-唾液酰化水平。A2,6-唾液酰化在UC小鼠中升高(圖2G)。與ST6GAL1+/+大鼠相比,ST6GAL1?/?大鼠在UC模型中表現出明顯的體重減輕(圖2H)。此外,UC造模后ST6GAL1?/-大鼠出現輕度大便不規則和便血。疾病活動指數(disease activity index, DAI)包括體重、大便形狀、便血等因素,常用于評估UC的嚴重程度。UC模型中,ST6GAL1-/-大鼠DAI評分低于ST6GAL1+/+大鼠(圖2I)。此外,UC模型中ST6GAL1+/+大鼠的結腸長度更短,而ST6GAL1?/?大鼠的結腸長度沒有變化(圖2J),表明ST6GAL1消融減輕了UC的癥狀。
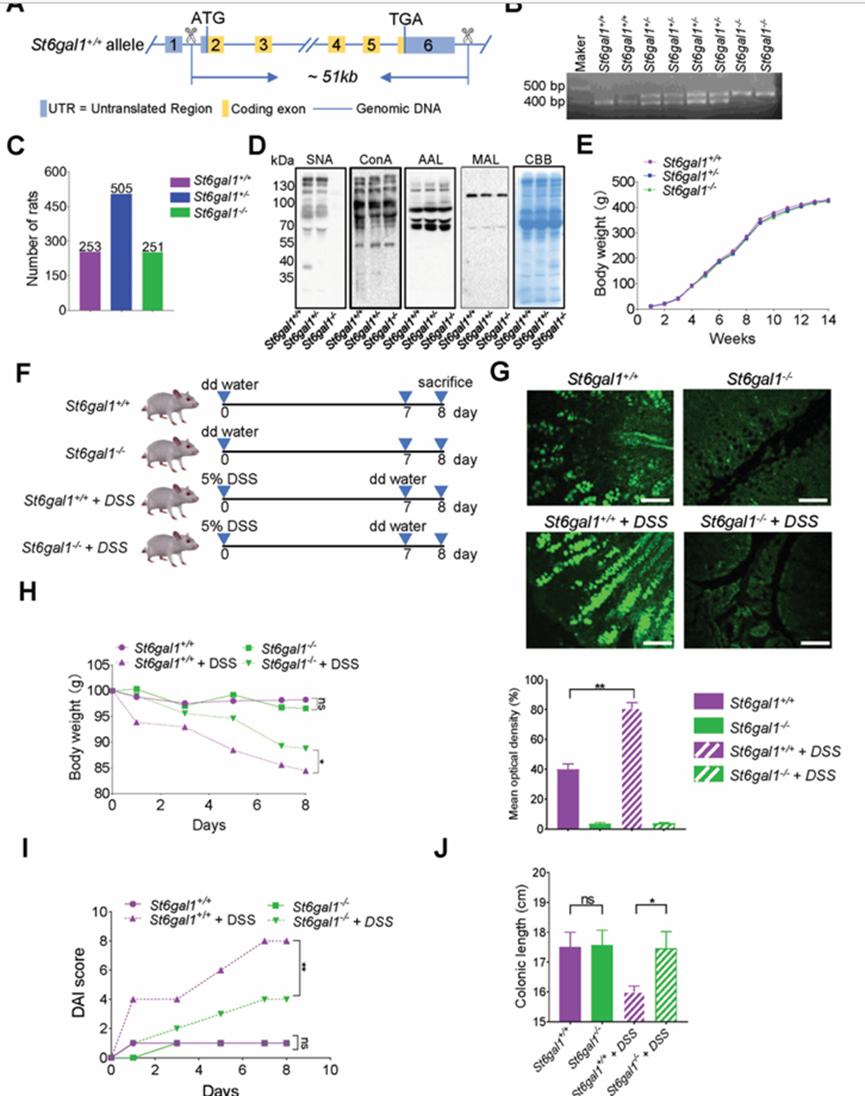
圖2、消融ST6GAL1可緩解UC大鼠UC癥狀
3、UC患者CD4+ t細胞中促炎細胞因子表達上調
蘇木精-伊紅染色(HE)顯示UC患者結腸損傷嚴重(圖3A),病理評分增高(圖3B)。此外,作者分離了ST6GAL1+/+和ST6GAL1?/?大鼠的腸上皮細胞,在體外用脂多糖(LPS)刺激腸上皮細胞,并表征了ST6GAL1的mRNA水平。2,LPS刺激后上皮細胞的6-唾液酰化增加。鑒于免疫失衡是UC發病的重要原因之一,作者利用單細胞測序技術和huARdb (https://huarc.net/數據庫)對UC患者的免疫細胞數量進行了分析。UC患者CD4+ T、Th1、Th17和Treg細胞數量增加,而Th2細胞數量減少(圖3C)。此外,結腸組織中CD4+ T細胞的數量急劇增加(圖3D)。基因本體(GO)富集分析也表明,UC患者的CD4+ T細胞和炎癥反應富集。UC組織中促炎因子如白細胞介素(IL)?2、IL-6、IL-17a和IFN- 水平顯著升高,而抗炎因子IL-4、IL-10和IL-13水平降低(圖3E)。接下來,作者測定了UC患者外周血中免疫細胞的數量。UC患者CD4+ T細胞數量顯著增高;然而,CD8+ T細胞數量未見變化(圖3F)。CD4+CD25+FoxP3+ (Treg)細胞數量減少(圖3G)。促炎因子il - 2、IL-6、IL-17水平顯著升高,抗炎因子IL-4、IL-10、IL-13水平降低(圖3H)。總體而言,UC患者的促炎細胞因子水平升高,并伴有CD4+ T細胞產生的抗炎細胞因子下調。
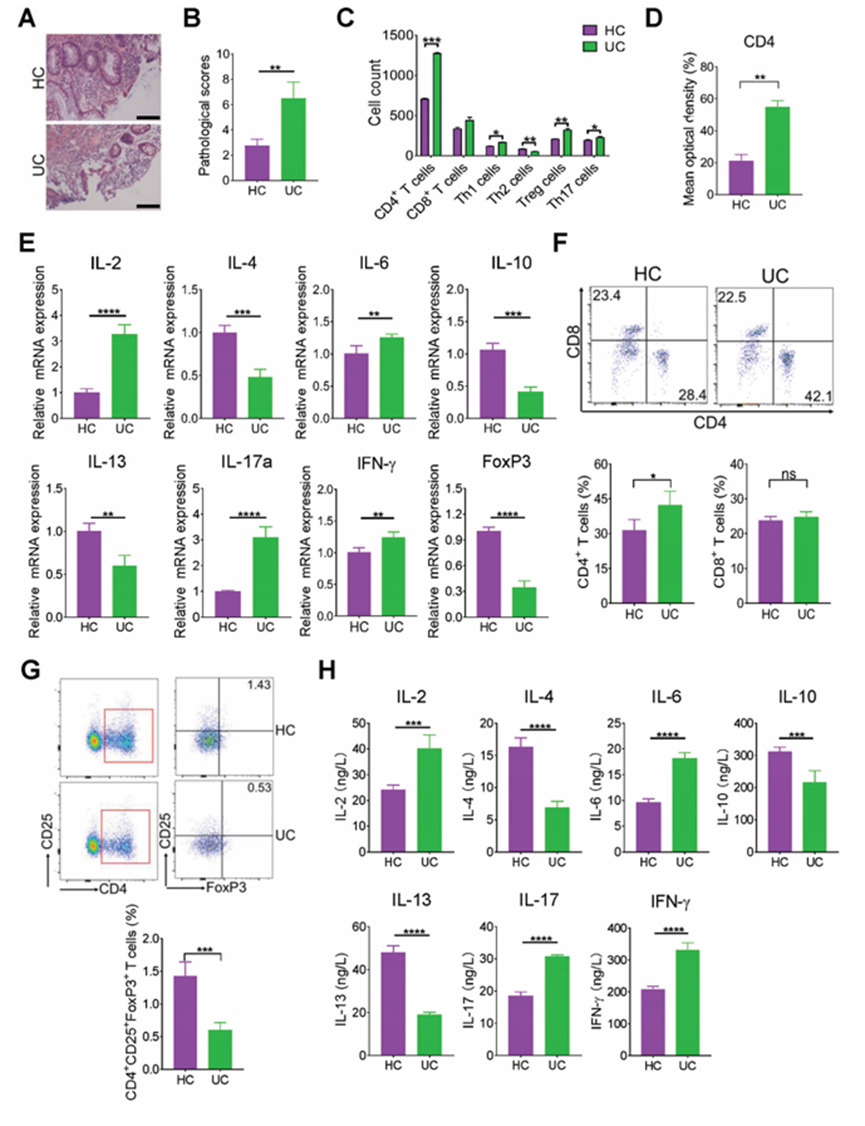
圖3、UC患者CD4+ t細胞數量和促炎細胞因子表達上調
4、ST6GAL1基因消融術減少UC大鼠CD4+ t細胞產生促炎細胞因子
ST6GAL1?/?UC大鼠UC癥狀得到緩解,UC患者CD4+ T細胞和促炎細胞因子水平顯著升高。這表明ST6GAL1影響CD4+ T細胞數量和細胞因子水平。因此,作者檢測了ST6GAL1+/+和ST6GAL1?/?UC大鼠的CD4+ T細胞數量和促炎細胞因子水平。ST6GAL1?/?UC大鼠的病理評分低于ST6GAL1+/+ UC大鼠(圖4A,B)。與ST6GAL1?/?UC大鼠相比,ST6GAL1?/?UC大鼠脾臟(圖4C)和Peyer’s patches(圖4D)中CD4+ T細胞數量減少。此外,與ST6GAL1?/?CD4+ t細胞相比,來自ST6GAL1+/+ UC大鼠的CD4+ t細胞促進了UC的發生。一些研究報道Treg和Th17細胞之間的平衡對維持腸道內穩態很重要。Treg細胞的減少或Th17細胞的增加會促進腸道炎癥。ST6GAL1?/?UC大鼠脾臟Treg細胞和Peyer 's斑塊數量增加,而Th17細胞數量減少。ST6GAL1?/?CD4+ T細胞中Treg細胞數量增加,而Th17細胞數量減少。此外,ST6GAL1?/?UC大鼠表現出低水平的促炎細胞因子IL-2、IL-6、IL-17a和IFN- 和高水平的抗炎細胞因子IL-4、IL-10和IL-13(圖4E)。上述結果表明,ST6GAL1消融可誘導CD4+ T細胞向treg細胞極化,抑制其向Th17細胞極化。
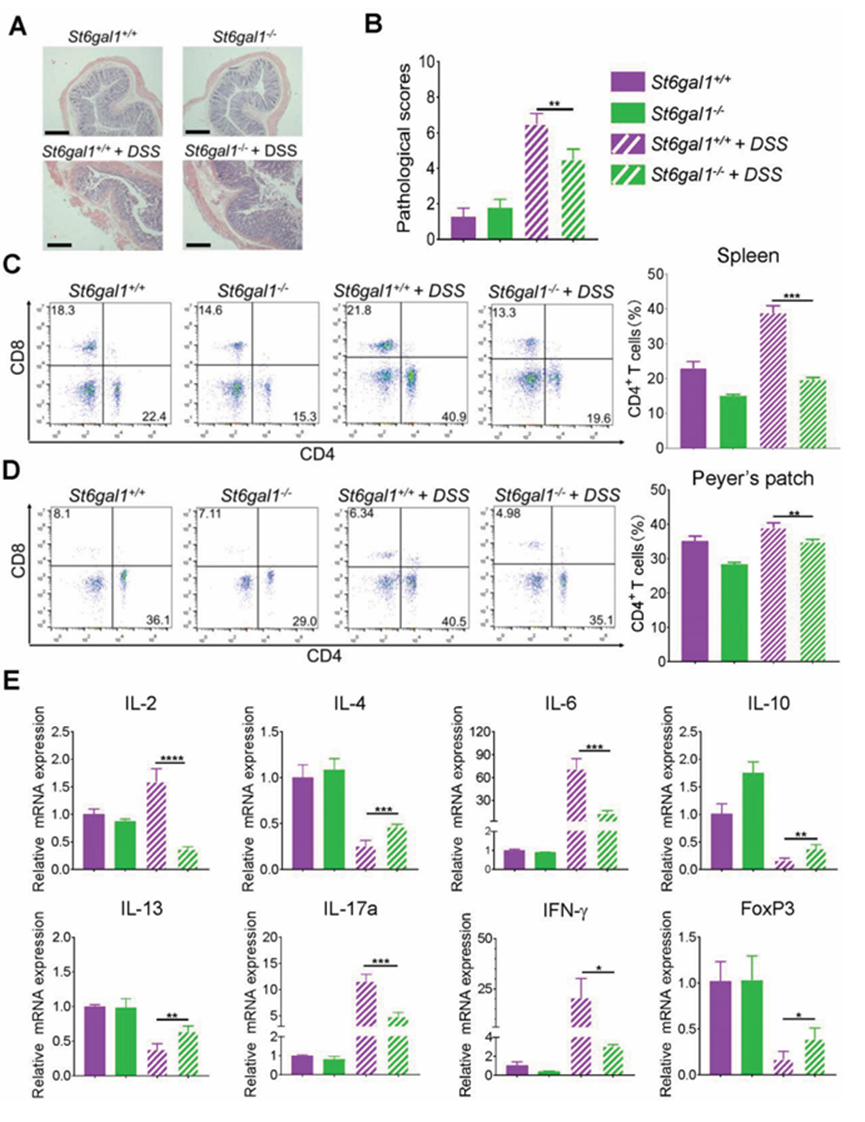
圖4、ST6GAL1基因消融術可減少UC模型大鼠CD4+ t細胞產生促炎細胞因子
5、ST6GAL1基因的消融術抑制CD4+ t細胞的增殖和活化
CD4+ T細胞的過度活化與UC的發病機制有關。為了研究ST6GAL1在CD4+ T細胞活化中的作用,作者采用磁珠分選方法從ST6GAL1 +/+和ST6GAL1?/?大鼠脾臟中分離CD4+ T細胞。在羧基熒光素二乙酸琥珀酰酯實驗中,ST6GAL1的消融抑制了抗CD3和CD28抗體刺激后的CD4+ t細胞(圖5A)。T細胞在激活后經常聚集成簇。與ST6GAL1+/+ CD4+ T細胞相比,激活后ST6GAL1?/- CD4+ T細胞聚集減少(圖5B)。流式細胞術還顯示,ST6GAL1?/-大鼠體內CD4+CD69+細胞(活化CD4+ T細胞)數量較少(圖5C)。為了進一步闡明ST6GAL1調控CD4+ T細胞活化的機制,作者建立了ST6GAL1基因敲低Jurkat細胞(Jurkat- shST6GAL1 cells), ST6GAL1恢復Jurkat- shST6GAL1 - re細胞。ST6GAL1 mRNA在Jurkat-shST6GAL1細胞中的表達降低,通過將ST6GAL1基因重新導入JurkatshST6GAL1細胞,ST6GAL1 mRNA的表達得以恢復(圖5D)。SNA凝集素印跡分析證實ST6GAL1基因表達水平。2,6-唾液化在Jurkat-shST6GAL1細胞中幾乎檢測不到,但在Jurkat-shST6GAL1- re細胞中恢復。ST6GAL1的缺失導致Jurkat細胞的增殖受到抑制,通過重新引入ST6GAL1, Jurkat細胞的增殖得以恢復(圖5E)。Jurkat-shST6GAL1細胞的細胞聚集被抑制,但在Jurkat-shST6GAL1- re細胞中恢復(圖5F)。在Jurkat-shST6GAL1細胞中,CD69+細胞的數量被抑制,并通過重新引入ST6GAL1恢復(圖5G)。
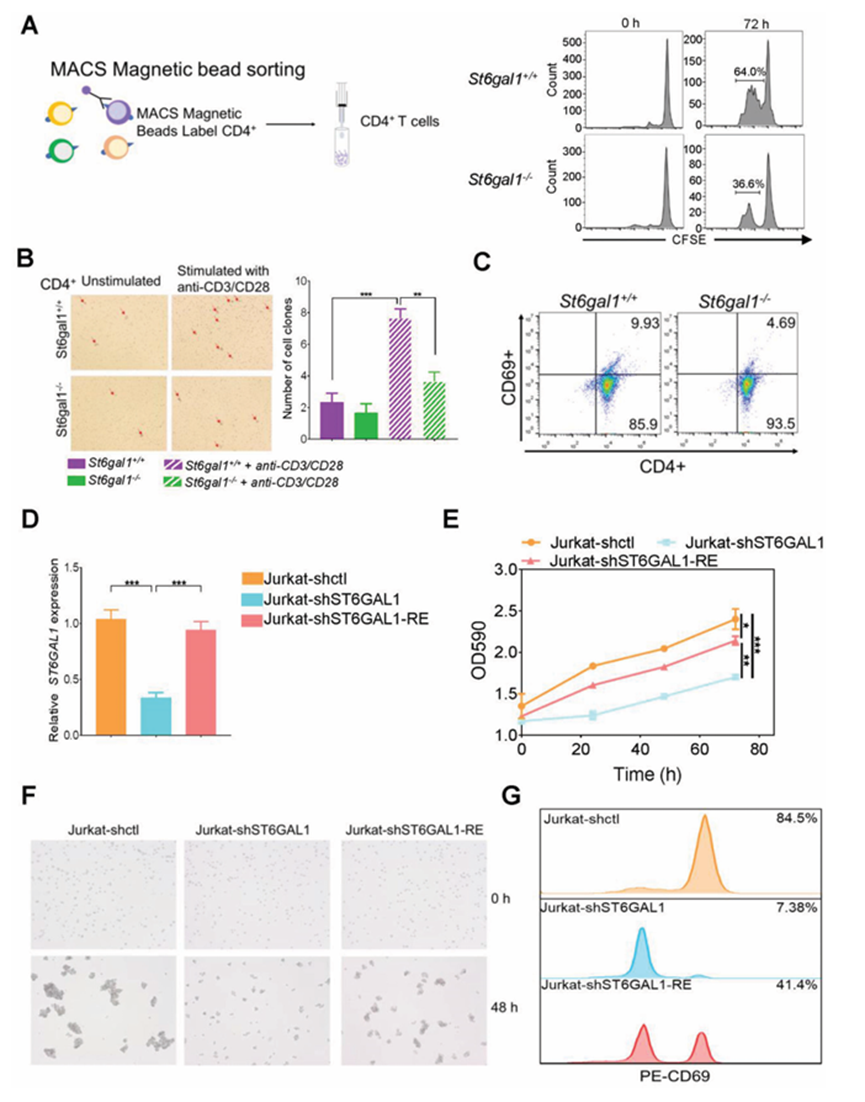
圖5、ST6GAL1基因在體外抑制CD4+ t細胞的增殖和活化
6、ST6GAL1基因的消融抑制t細胞受體(TCR)向脂筏的易位并減弱CD4+ t細胞中TCR信號傳導
在對t細胞激活的反應中,TCR與脂質筏相關,脂質筏作為信號傳導和運輸的平臺。作者首先使用質譜法測定了CD4+ T細胞TCR的n -聚糖譜。在TCR中鑒定出含有單、二、三或四半乳糖的唾液化n -聚糖。在ST6GAL1?/- CD4+ t細胞中,TCR上n -聚糖的2,6-唾液酰化被消除。用共聚焦顯微鏡觀察抗CD3/CD28刺激后CD4+ T細胞表面TCR的分布。Flotillin-1是脂筏的標志物,對脂筏的結構和功能有重要影響。ST6GAL1的消融抑制了脂筏的形成,Flotillin -1的低表達證明了這一點(圖6A,B)。與ST6GAL1+/+ CD4+ T細胞相比,ST6GAL1?/- CD4+ T細胞中脂筏中TCR的水平降低,重新引入ST6GAL1可以恢復這些影響(圖6A)。作者檢測了抗CD3 /CD28刺激后的TCR信號通路。ST6GAL1基因的消融抑制了TCR下游的Zap70、p38和細胞外調節蛋白激酶(ERK)的磷酸化(圖6C)。NF-B (p65)在ST6GAL1?/- CD4+ t細胞中的表達下調(圖6C)。此外,ST6GAL1的缺失抑制了p-p65在CD4+ T細胞中的核易位。NF-B的激活往往會誘導促炎細胞因子的產生,從而誘發UC,因此作者分析了NF-B與ST6GAL1之間的相互作用。染色質免疫沉淀-實時聚合酶鏈反應(ChIP-qPCR)顯示NF-B可以結合ST6GAL1啟動子上游的- 129至- 109堿基對(bp)。與ST6GAL1+/+ CD4+ T細胞相比,在ST6GAL1?/- CD4+ T細胞中,NF-B與ST6GAL1的相互作用被抑制(圖6D)。為了進一步研究ST6GAL1在T細胞活化過程中的作用,作者使用轉錄組學分析了ST6GAL1+/+和ST6GAL1?/- CD4+ T細胞中的基因表達。與ST6GAL1+/+ CD4+ T細胞相比,ST6GAL1?/?CD4+ T細胞顯示555個下調基因和551個上調基因。氧化石墨烯分析顯示,ST6GAL1的缺失抑制CD4+ T細胞激活和炎癥反應(圖6E)。京都基因與基因組百科全書(KEGG)分析顯示,ST6GAL1的缺失下調了多個信號分子的表達,如TCR、NFB、IL-17、TNF和PI3K-AKT(圖6F)。相反,組織轉錄組學分析顯示,UC患者的TNF、IL-17、NF-B和PI3K-AKT通路增強。這些結果表明,ST6GAL1通過調節TCR、NF-B等信號通路影響CD4+ T細胞的活化。ST6GAL1的缺失抑制了Jurkat-shST6GAL1細胞中TCR的表達和脂質筏的形成,這種抑制可以通過將ST6GAL1重新引入Jurkat-shST6GAL1細胞來恢復。免疫印跡結果與免疫熒光結果一致。在Jurkat-shST6GAL1細胞中,ST6GAL1的缺失抑制了Zap70、p38和ERK的磷酸化,并下調了NF-B的表達,這些變化在Jurkat-shST6GAL1- re細胞中得以恢復。CHIP-qPCR分析顯示,NF-B在Jurkat細胞系和CD4+ T細胞中與ST6GAL1啟動子上游區域的- 129至- 109 bp位置結合。作者通過轉錄組學分析分析ST6GAL1基因對Jurkat細胞基因表達譜的影響。與Jurkatshctl細胞相比,850個基因表達上調,1590個基因表達下調。氧化石墨烯分析顯示,ST6GAL1基因敲低可降低T細胞活化、細胞表面受體信號傳導、炎癥反應和其他生物學功能KEGG分析顯示,ST6GAL1基因沉默降低了TCR、TNF-、IL-17、細胞外基質(Extracellular Matrix, ECM)和其他信號通路。
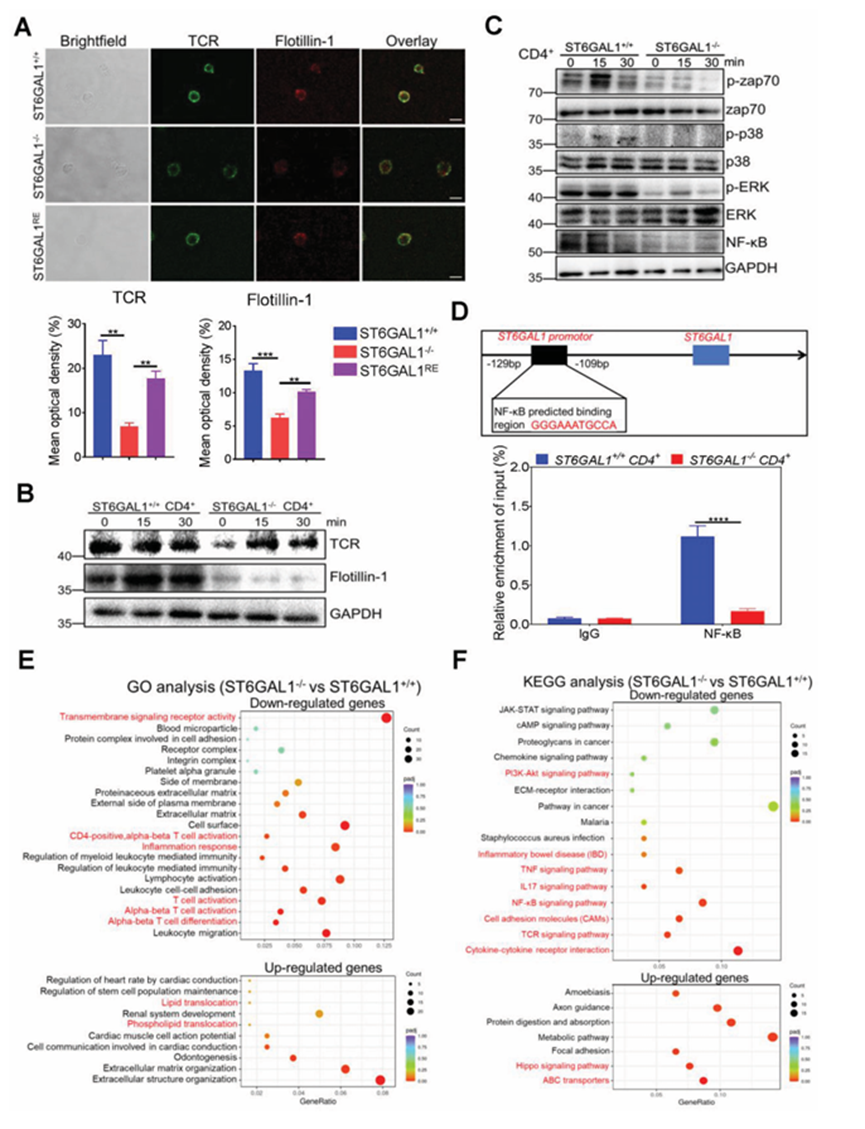
圖6、ST6GAL1基因的消融可抑制TCR向脂筏的易位,并減弱CD4+ t細胞中的TCR信號傳導
7、ST6GAL1與NF-B在UC發病中呈正相關
為了研究ST6GAL1與NF-B在UC發病機制中的相關性,作者檢索了GEO數據庫中GSE179285的數據。UC患者結腸中ST6GAL1和NF-B的表達水平較HCs升高(圖7A)。Spearman相關分析顯示,UC和HCs患者中ST6GAL1與NF-B呈正相關。為了進一步驗證NF-B與ST6GAL1的相關性,作者采用RT-qPCR檢測了13名健康志愿者和14名UC患者結腸組織中NF-B和ST6GAL1的mRNA水平。UC組NF-B和ST6GAL1的表達明顯升高(圖7B),二者表達呈正相關(圖7C、D)。作者使用雙熒光素酶報告試驗進一步驗證了ST6GAL1和NF-B之間的相互作用。NF-B結合到ST6GAL1基因的啟動子區域并啟動轉錄,導致熒光素酶活性顯著增加(圖7E),表明NF-B與ST6GAL1基因表達呈強正相關。此外,NF-B和ST6GAL1的表達與UC的嚴重程度呈正相關(圖7F,G),這表明NF-B和ST6GAL1抑制劑的開發可能有助于治療UC。
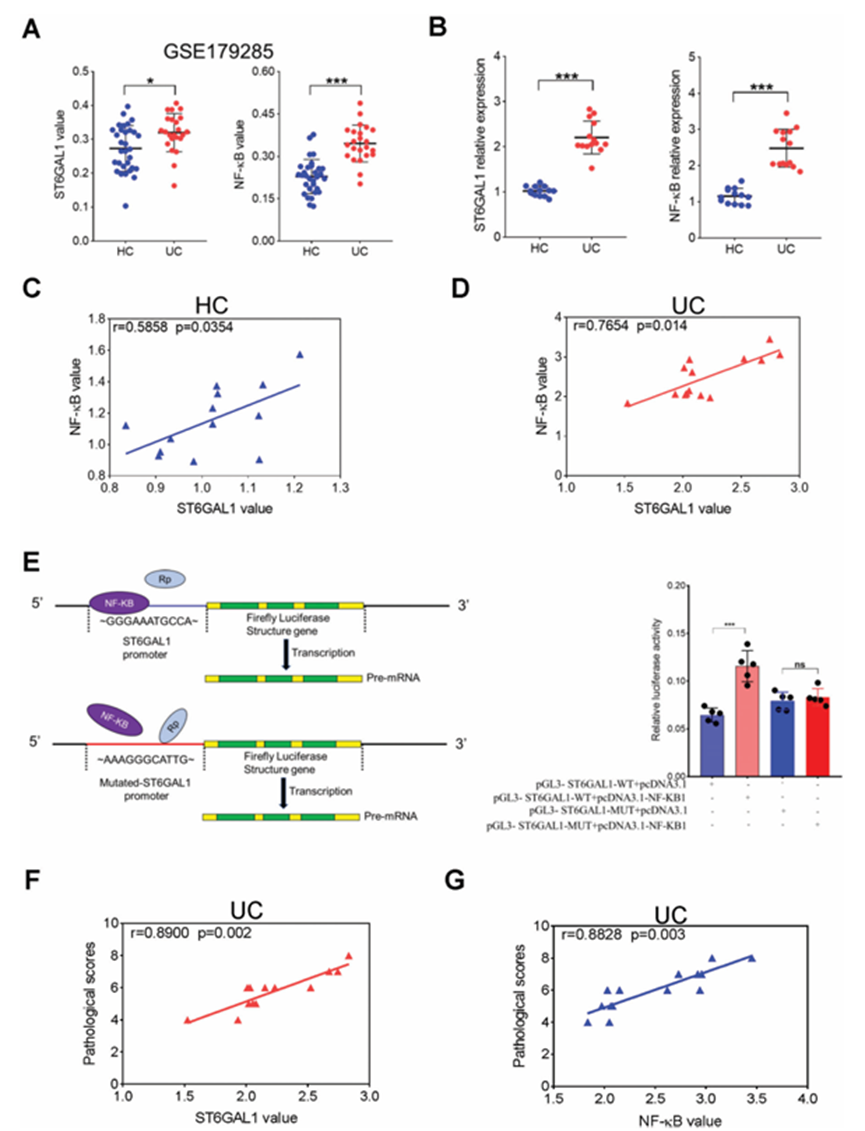
圖7、ST6GAL1在UC發病過程中與NF-B呈正相關
結論
T細胞活化伴隨著糖基轉移酶、糖苷酶及其底物的差異表達,這些差異在調節t細胞應答中起著直接而有力的作用。ST6GAL1缺乏可抑制TCR的2,6-唾液化,抑制TCR進入CD4+ t細胞脂筏,減弱TCR信號強度,進一步下調NF-B的表達,減少促炎細胞因子的產生,減輕UC。ST6GAL1可調節多種糖蛋白的生物學功能;因此,2,6-唾液化蛋白的廣泛變化使得很難確定ST6GAL1在單個糖基化蛋白激活T細胞中的作用。然而,我們的觀察結果顯示UC的發病機制部分負責與CD4+ T細胞中ST6GAL1高表達相關的功能改變,這是UC臨床治療的潛在新靶點。
實驗方法
CRISPR-CAS9、慢病毒轉染、免疫熒光、流式細胞術、雙熒光素酶報告基因測定
參考文獻
Fan Q, Li M, Zhao W, Zhang K, Li M, Li W. Hyper α2,6-Sialylation Promotes CD4+ T-Cell Activation and Induces the Occurrence of Ulcerative Colitis. Adv Sci (Weinh). 2023 Sep;10(26):e2302607. doi: 10.1002/advs.202302607. Epub 2023 Jul 9. PMID: 37424034; PMCID: PMC10502867.