






人胰島的單核 RNA 測序識別新基因組 并區分具有動態轉錄組概況的 β 細胞亞群
該研究于2023年5月發表......
單細胞 RNA 測序 (scRNA-seq) 為人類胰島細胞類型及其相應的穩定基因表達譜提供了寶貴的見解。然而,這種方法需要細胞解離,這使其在體內的應用變得復雜。另一方面,單核 RNA 測序 (snRNA-seq) 與冷凍樣品兼容,消除了解離誘導的轉錄應激反應,并提供了可用于識別前體 mRNA 轉錄本的內含子序列的增強信息。作者使用snRNA-seq分析顯示人胰島內分泌細胞體外和體內差異選擇性表達前四位的基因不是經典基因,而是一組新的非經典基因標記,包括ZNF385D、TRPM3、LRFN2、 PLUT(β細胞); PTPRT、FAP、PDK4、LOXL4(α-細胞); LRFN5、ADARB2、ERBB4、KCNT2(δ 細胞);和 CACNA2D3、THSD7A、CNTNAP5、RBFOX3(γ 細胞)。其次,通過整合來自人胰島細胞的 scRNA-seq 和 snRNA-seq 的信息,我們區分了三個 β 細胞亞群:INS pre-mRNA 群 (β3)、中間 INS mRNA 群 (β2) 和 INS富含 mRNA 的簇 (β1)。這些顯示不同的基因表達模式,代表體外和體內不同的生物動態狀態。有趣的是,富含 INS mRNA 的簇 (β1) 成為體內主要的亞簇。綜上所述,人胰島細胞的 snRNA-seq 和 pre-mRNA 分析可以準確識別人胰島細胞群、亞群及其體內動態轉錄組概況。
該研究于2023年5月發表發表在《Genomemedicine》,IF:15.266。
技術路線:

實驗方法:人胰島細胞和細胞核加工、人胰島細胞移植、scRNA-Seq、snRNA-Seq、無監督數據分析、偽時間分析和RNA速度分析、通路分析、RNA原位雜交。
1、人胰島細胞和細胞核的RNA-seq分析
圖1A和B描述了用于這些研究的方法和數據分析工作流程。
作者分析了10,732個細胞和11,018個核。與scRNA-seq方法(圖1C)相比,snRNA-seq方法中每個細胞的基因和讀數測序的數量較低。在細胞核中測序的線粒體基因的TE百分比低于1%,明顯低于在細胞制備中測序的線粒體基因(圖1C)。在snRNA-Seq數據中,當分析內含子加外顯子讀取時,基因和讀取的數量明顯高于單獨的外顯子讀取(圖1D)。
基于上述數據質量,我們接下來將使用外顯子讀取的scRNA-seq與使用內含子和外顯子讀取的snRNA-seq進行比較,以改進基因檢測和映射。scRNA-seq分析核轉錄本和細胞質轉錄本,其中大部分是細胞質轉錄本,而snRNA-seq分析的主要是核轉錄本,而在細胞核分離過程中,來自細胞質或粗糙ER的轉錄本最少。因此,我們預計,在scRNA和snRNA測序過程中,RNA-seq讀數會不同。在細胞中,17±0.7%的讀數為內含子讀出,而在細胞核中為53±1.9%。另一方面,細胞中73±1.6%的讀出為外顯子讀出,而細胞核中為32±0.8%。因此,只有當核與核或細胞與細胞進行比較時,基因表達相關性才會完全或接近完全線性(圖1E),而當細胞與細胞核相比時,這種相關性較低(圖1F)。
兩種RNA測序方法(UMI>20)檢測到的共同基因數量為16,452個(71.3%),而1433個基因(6.2%)僅在scRNA-seq中檢測到,5184個基因(22.5%)僅在snRNA-seq中檢測到(圖1G)。表明兩種方法檢測到的幾乎29%的基因對于相同的人胰島樣本是不同的,兩種RNA測序方法可能揭示了胰島細胞群體的同一性。
有趣的是,用Seurat的整合算法,使用外顯子讀數(scRNA-seq)或內含子加外顯子讀數(snRNA-seq)對細胞和細胞核進行無監督聚類,揭示了具有相似位置且在其UMAP中具有強烈重疊的簇(圖1H,I)。
2、使用scRNA-seq和snRNA-seq對人類胰島細胞類型進行監督分類
我們將scRNA-seq和snRNA-seq數據集投影到一個公開可用的Azimuth整合人類胰腺參考文獻中,該參考文獻包含六個不同的scRNA-seq數據集,這些數據集是使用Seurat的幾種不同單細胞技術生成的(圖1B和2A-D)。投影是通過scRNA-seq的外顯子讀數(圖2A、B)和snRNA-seq的外顯子或內含子加外顯子讀數(圖2C、D)完成的。當我們將當前研究的人類胰島scRNA-seq數據投影到參考上時,我們發現不同的人類胰島細胞群幾乎完美對齊,預測分數中位數為1,平均值為0.948(圖2A,B)。使用外顯子讀數或內含子加外顯子讀數將snRNA-seq數據投影到參考也導致高度對齊(圖2C,D),外顯子加內含子的預測分數顯著高于單獨外顯子(圖2D)。為了確定觀察到的與外顯子加內含子讀取的參考比對的更高概率是否可以用不同的UMI深度來解釋,我們對包含內含子的庫進行二次采樣,中值為1247 UMI(外顯子只讀庫的中值)并分析了參考對齊預測分數。我們發現在二次采樣后,包含內含子的文庫的比對預測分數仍然優于僅包含外顯子的文庫。這進一步驗證了使用來自內含子和外顯子讀取的信息,并使用snRNA-seq對人類胰島細胞群進行詳細分析。數據還表明,來自scRNA-seq和snRNA-seq的人類胰島細胞簇具有高度相似性,并且用于鑒定人類胰島細胞類型簇時,包含內含子和外顯子讀數的snRNA-seq數據可與scRNA-seq數據可以互換。
接下來,我們測試了從scRNA-seq和snRNA-seq數據(圖2A、C)生成的不同簇與不同內分泌細胞簇中典型基因的基因表達水平的關聯(圖2E-H)。在scRNA-seq中觀察到已建立的典型基因細胞標記(GCG、INS、SST和PPY)與其他幾種已知選擇性標記與α-、β-、δ-和γ-細胞之間的強相關性。因此,在scRNA-seq分析中相應地分配了UMAP中的細胞類型(圖2E,G)。然而,在scRNA-seq中觀察到的這些典型基因標記的強相關性在snRNA-seq中對于α-、β-和γ-細胞較弱(圖2F,H)。例如,在β細胞中,INS和IAPP是差異表達最多的基因,而在snRNA-seq數據集中它們不是差異表達最多的基因。這些結果強調需要鑒定新的基因組作為胰島內分泌細胞的標記,以便在snRNA-seq分析中更恰當地定義它們。

3、用于鑒定人胰島內分泌細胞類型的snRNA-seq數據集中的新基因集
在scRNA-seq和snRNA-seq樣本之間進行了差異基因表達分析,并研究了snRNA-seq富集基因的生物型。即使包含內含子讀數,snRNA-seq中的大多數基因都是蛋白質編碼基因(圖3A)。接下來,注釋了snRNA-seq數據,并投影到Azimuth的人類胰腺參考上,并用整個數據集測試了每個簇的差異基因表達(圖1B)。使用這種方法,每個細胞簇中差異表達的基因被鑒定為p值~0和log2FC大于1.5。即使候選基因符合這些標準,我們也忽略了在其他細胞簇中表現出相當大的表達(log2FC>0.85)的基因。因此,制作了一個表,其中包含每種細胞類型的前四個差異表達基因(圖3B)。有趣的是,scRNA-seq中定義內分泌細胞的前四個差異表達基因(即INS、GCG、STS或PPY)都沒有出現在snRNA-seq中差異和選擇性表達基因的簡短列表中。這表明這些典型基因不是snRNA數據集中在α-、β-、δ-和γ細胞中差異表達最高的基因。為了確認這些新鑒定的內分泌細胞基因標記的可靠性,我們在snRNA-seq和scRNA-seq數據對象上測試了它們。它們在snRNA-seq和scRNA-seq的相應細胞簇中顯示出清晰且主要是排他性的表達模式,但在snRNA-seq數據集中具有更高的表達(圖3C,D)。
前幾個內分泌細胞基因標記顯示出比其相應的典型單細胞聚類基因標記更具特色的定位模式,分別用于snRNA-seq數據對象中的α-、β-、δ-和γ-細胞(圖2H和3E)。值得注意的是,CNTNAP5在γ細胞中沒有表現出高度不同的表達模式,但它被認為是基于高調整p值(1.58×10-5)和log2FC為1.654的γ細胞基因標記。有趣的是,在非內分泌細胞的snRNA-seq分析中,差異表達最高的基因包含在scRNA-seq中定義這些細胞類型的典型基因標記(REG1A、CFTR、FLT1、COL1A1和PRKG1,用于腺泡、導管、內皮、分別為激活的星狀細胞、靜止的星狀細胞)。這表明人類內分泌細胞和非內分泌細胞之間關于規范基因的穩態轉錄本豐度的有趣二分法(圖3F)。
為了驗證這些來自snRNA-seq分析的新鑒定的基因作為細胞標記的存在,我們聚焦于β細胞,并使用RNA顯微鏡檢測來自健康捐贈者的分散的人胰島細胞中的ZNF385D mRNA。如圖3G所示,ZNF385D mRNA在人β細胞中明顯且唯一地被檢測到。使用內含子探針的表達僅限于細胞核(圖3G,上圖),而使用外顯子探針的信號定位在細胞質和細胞核中(圖3G,下圖)。

4、比較scRNA-seq和snRNA-seq分析在區分三種不同的β細胞亞型時的異同
為了識別β細胞簇中的人類β細胞亞型,我們創建了一個新的數據對象,集成了scRNA-seq和snRNA-seq數據集,以比較INS基因表達模式(圖1B和4A-C)。結合這兩個數據集有效地增加了分析的能力,同時提供了來自細胞質和核轉錄組的額外信息。在對已識別的β細胞簇進行子集化后,我們生成了Louvain分辨率為0.8的簇(圖4C)以分配三個β細胞亞簇。scRNA-和snRNA-seq數據對象之間的β細胞簇中的INS基因表達在拓撲位置方面是不同的(圖4D、E)。聚類3在scRNA-seq數據中顯示較低的INS表達,但在snRNA-seq數據對象中顯示最高(圖4E),這表明聚類3包括主要具有INS pre-mRNA的β細胞,因為snRNA-seq主要分析pre-mRNA。
接下來,我們創建了偽時間和RNA速度軌跡圖,將聚類3作為基礎(圖4F),并根據偽時間或RNA速度軌跡重新排列了每個聚類的順序,分為β1、β2和β3細胞(圖4G)。使用兩個數據集的綜合數據進行偽時間或RNA速度分析時,從聚類3到聚類1的類似進展發生。單個scRNA-seq數據的RNA速度分析顯示了從聚類3到聚類1的類似轉變。有趣的是,僅使用snRNA-seq數據的RNA速度分析顯示從聚類3末端到聚類3邊緣的分叉,然后從聚類2到聚類1。這些結果表明,可以通過綜合或單個RNA-seq數據觀察到β細胞亞聚類的識別和轉變,但是這種轉變似乎在scRNA-seq和snRNA-seq之間有所不同。這很可能是因為RNA速度基于外顯子-內含子讀數比率,利用新轉錄的未剪接mRNA推斷基因表達狀態的時間導致,但是snRNA-seq中外顯子-內含子讀數較低,使得在聚類之間的轉變的解釋變得復雜。因此,對于綜合或scRNA-seq數據,RNA速度是適當的,但是對于snRNA-seq,似乎RNA速度不太適合分析β細胞成熟的進展。
由于β1細胞簇中的細胞具有從scRNA-seq(成熟mRNA)β細胞簇對象推斷出的穩定INS表達,但從snRNA-seq(pre-mRNA)β細胞簇對象推斷出非常低的INS表達,我們將這些細胞視為富含INS的細胞亞群。接下來,我們觀察了ZNF385D表達,它是β細胞中snRNA-seq中差異表達最高的基因,發現scRNA-seq數據對象中β細胞亞群中的表達最小,但snRNA-seq數據中的拓撲位置不同對象(圖4H,I),其中β1代表具有高ZNF385Dpre-mRNA表達的細胞,與INS表達相反(圖4E)。接下來,我們觀察了INSmRNA結合蛋白HNRNPA2B1的表達,這是一種調節INSmRNA穩定性和翻譯的RNA結合蛋白,以確定β2細胞簇是否代表β3和β1之間的過渡階段。如圖4J、K所示,與聚類3相比,聚類2和聚類1中的HNRNPA2B1表達增加,表明聚類2可能代表“轉變中”β細胞類型,其中細胞從轉錄活性β3細胞轉變為β1細胞具有成熟儲存的INS mRNA。

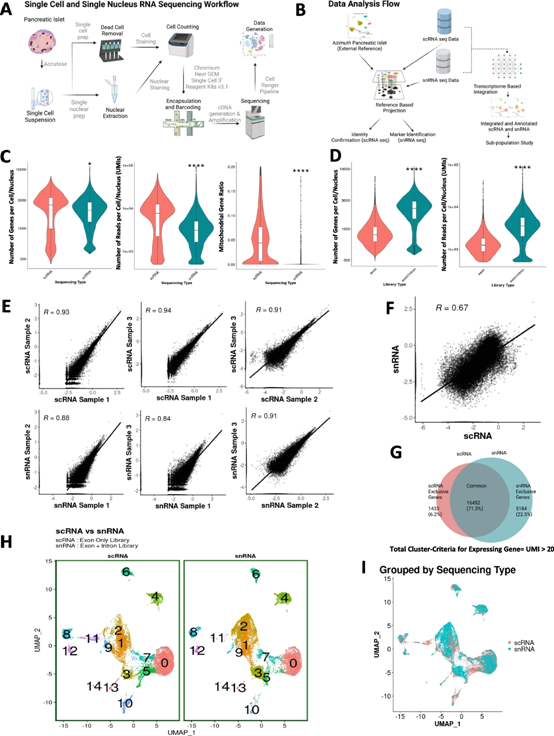
5、三種不同β細胞亞型的基因通路分析
為了研究β3、β2和β1之間潛在的生物學差異,我們對來自scRNA和snRNA-seq實驗的組合數據集進行了GSEA。有趣的是,定義細胞外基質(ECM)形成、相互作用和反應的生物過程的基因富集獨特地存在于β3亞群中(圖5A)。同樣,GSEA也表明細胞內囊泡出芽、運輸和形成的生物學過程主要存在于β2亞群中(圖5B),而胰島素分泌、加工和葡萄糖代謝生物學過程的基因主要存在于β2亞群中。在β2和β1子簇中表示(圖5C)。當我們將來自兩種RNA-seq方法的數據集輸入GSEA時,還獲得了關于對β細胞的發育、分化、基因表達調控和增殖具有重要意義的幾個生物學過程的附加信息。
如圖5D所示,參與β細胞增殖的基因在β3亞群中表達更高,參與基因表達調控的基因在β2亞群中突出,而參與β-細胞發育和分化在β1亞群中表達更高。這些結果清楚地描述了不同β細胞亞群中特定細胞功能的不同基因集,強調了人類胰島中β細胞的異質性。
6、人體內胰島的snRNA-seq分析
接下來,我們試圖使用移植到血糖正常的免疫抑制小鼠體內的人類胰島移植物來檢查體內不同的人類胰島細胞群。為此,我們將來自四名健康供體的1000個人類IEQ移植到RAG1-/-小鼠的腎囊中,并在移植后3個月收獲移植物(圖6A)。
通過Minute?單核分離試劑盒直接從移植物中提取細胞核,在質量評估和計數后將每個樣本5000-16,000個細胞核加載到10X GenomicsChromiumController中,poly-A轉錄本逆轉錄擴增,cDNA標記,以及得到的文庫測序深度為每個樣本250-5億個讀數。最終,分析了7765個細胞核。基因數為2226±263,基因計數為4125.64±263,決定測序效率的可用讀數與測序讀數之比為0.999±0.001。在細胞核制備物中測序的線粒體基因百分比低于1%。snRNA-seq數據被投射到Azimuth人類胰腺參考上,以使用規范基因集以及之前描述的新基因集(圖1A和6A)識別胰島細胞群。此外,snRNA-seq數據被投影到來自體外研究的集成scRNA-seq和snRNA-seq數據上,用于分析不同的β細胞亞群(圖6A)。去除基因計數和/或UMI計數異常值或線粒體基因高表達或環境RNA污染≥20%的細胞。在質量控制過程中,我們發現≤10%的小鼠基因比率對于在這種胰島移植環境中識別體內不同細胞類型是最佳的,而小鼠基因對聚類模式沒有顯著影響。
使用Azimuth的基于參考的縮減和注釋,我們確認了七個不同的人類胰島細胞簇,主要由內分泌細胞組成(圖6B)。為簡單起見,我們從數據集中省略了少于五個細胞的簇。通過體外snRNA-seq分析鑒定的基因標記集(圖3C)在基于平均表達的點圖(圖6C)和散點圖(圖6D)與典型基因標記的非特異性模式相比。當使用基于內部參考的減少和注釋時,體外snRNA-seq分析中新鑒定的基因集的這種特異性細胞簇比對持續存在(圖6E-G)。

7、人體內胰島中的β細胞亞型
首先,我們確定在scRNA-seq/snRNA-seq體外研究中鑒定的β細胞亞型是否在體內環境中仍然存在于人類胰島中。因此,我們將體內snRNA-seq數據集投影到體外scRNA-/snRNA-seq數據集參考上,該數據集預先標記了β細胞亞型(圖6E)并提取了β細胞簇(圖7A)。我們從主要數據簇中分離出β細胞亞簇并檢查基因表達模式(圖7A-H)。有趣的是,與體外人胰島的snRNAseq數據相反,體內人胰島中的β1簇與β2和β3簇相比顯示出相似水平的INS表達(圖7B,C)。當分析先前來自人類胰島移植物的snRNA-seq數據集時,觀察到類似的結果。此外,在我們的數據集中,ZNF385D和HNRNPA2B1的表達在體內和體外人胰島的β細胞亞群中似乎相似(圖7D-G)。重要的是,與體外相比,體內人胰島中β1亞群中具有穩定INS表達的細胞比例顯著增加,而β2轉換群和主要含有INS pre-mRNA的β3細胞比例顯著降低(圖7H)。我們推測這可能代表細胞從較低的INS表達β3和β2組到更成熟的INSmRNAβ1組的表型或成熟轉移,與生理性較低的體外條件相比,可能反映了體內微環境的成熟特征。
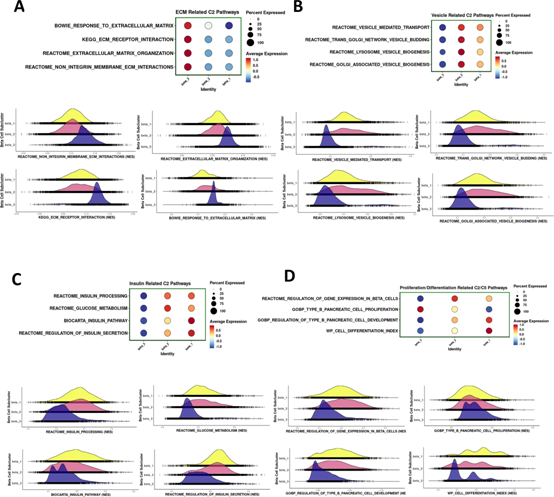
我們還使用與上述相同的方法對體內人類胰島的不同β細胞亞群進行了基因集富集分析(圖5和圖8)。正如在體外觀察到的(圖5),與β2和β1細胞亞型相比,β3細胞亞型顯示出更高水平的參與ECM生物過程的基因表達(圖8A)。在β2和β1細胞亞型中,參與細胞內囊泡出芽、運輸和形成、胰島素分泌、加工和葡萄糖代謝、β細胞分化、發育、增殖和基因表達調控等生物學過程的基因表達在體內和體外保持相當(圖8B-D)。值得注意的是,人類β3細胞體內與體外β細胞增殖相關的基因表達低于體外(圖8B-D),再次表明體內微環境可能為β細胞提供線索以支持功能更強、但與生理性較低的體外環境相比,有絲分裂狀態較低。


參考文獻:
Kang,R.B.,Li,Y.,Rosselot,C.etal.Single-nucleusRNAsequencingofhumanpancreaticisletsidentifiesnovelgenesetsanddistinguishesβ-cellsubpopulationswithdynamictranscriptomeprofiles.GenomeMed15,30(2023).https://doi.org/10.1186/s13073-023-01179-2