






全基因組CRISPR篩選發現DOCK1抑制和二甲雙胍在肝癌中的合成致命性
二甲雙胍是多種癌癥強有力的候選抗腫瘤藥物。然而它的抗腫瘤效果在不同的癌癥或亞群中有所不同,這可能是由于腫瘤的異質性。目前尚不清楚哪些肝細胞癌(HCC)患者亞群可以從二甲雙胍治療中受益。通過基于CRISPR-Cas9的全基因組敲除篩選,我們發現DOCK1水平決定了二甲雙胍的抗腫瘤作用,并且DOCK1是二甲雙胍在HCC中的合成致死靶點。在機制上,二甲雙胍促進DOCK1磷酸化,激活RAC1促進細胞存活,導致二甲雙胍耐藥。DOCK1選擇性抑制劑TBOPP通過二甲雙胍增強體外肝癌細胞系和患者來源的HCC類器官的抗腫瘤活性,并在體內異種移植的肝癌細胞和免疫能力強的小鼠肝癌模型中增強抗腫瘤活性。二甲雙胍可以改善DOCK1低水平HCC患者的總生存期,但對DOCK1高表達患者沒有改善作用。這項研究表明,二甲雙胍的有效性取決于DOCK1的水平,二甲雙胍聯合DOCK1抑制可能為二甲雙胍耐藥的HCC患者提供一種有前途的個性化治療策略。本文于2022年11月發表于Protein&Cell(IF=15.328)。
技術路線:

結果:
(1) CRISPR-Cas9文庫篩選確定DOCK1為二甲雙胍敏感性的決定因素
為了系統地識別對二甲雙胍治療敏感的肝癌亞型,我們采用了基于CRISPR-Cas9的負選擇方法來篩選那些損失會增強二甲雙胍抗腫瘤作用的基因。在二甲雙胍存在或不存在的情況下,用含有基因組級CRISPR敲除文庫(GeCKOv2)的慢病毒轉染的PLC/PRF/5細胞(PLC)進行培養。孵育兩周后,基因組DNA被分離出來,高通量測序用于確定引導RNA的豐度,然后用MAGeCKFlute進行進一步分析(圖1A)。然而,在二甲雙胍治療組中,398個基因顯著減少(差異beta評分<?0.3)(圖1B)。為了確定哪些基因可能使PLC細胞對二甲雙胍敏感,但在未處理的細胞中沒有表現出明顯的生長障礙,我們使用對照組β-評分變化不超過0.15(?0.15<對照組β-評分<0.15)的附加標準縮小了候選基因。根據這一標準,398個基因中有171個被確定為候選基因(圖1C),最終選擇其中6個進行進一步分析。
接下來,為了驗證篩選結果,我們定量了這6個候選細胞的mRNA表達,并測定了二甲雙胍在9種肝癌細胞中的半最大抑制濃度(IC50)值(圖1D和1E)。相關分析顯示DOCK1表達與二甲雙胍IC50值和AUC評分的Pearson相關系數最高(圖1F),提示DOCK1表達水平可能決定了肝癌細胞對二甲雙胍的敏感性。考慮到高差異beta評分及其與二甲雙胍反應的強相關性,我們將重點放在DOCK1上進行進一步研究。
對指導RNA的分析顯示,在二甲雙胍處理的細胞中,所有六種靶向DOCK1的sgRNAs的豐度都較低(補充圖未展示)。與這些結果一致,DOCK1敲低導致PLC細胞在長期集落形成實驗和IC50檢測中顯著敏化,以分析短期細胞活力(圖1G)。DOCK1的異位表達減弱了shDOCK1誘導的PLC細胞的二甲雙胍敏感性(圖1H)。此外,DOCK1的過表達消除了二甲雙胍在Huh7細胞中的抗腫瘤作用(圖1I)。綜上所述,這些結果表明DOCK1的表達水平決定了肝癌細胞對二甲雙胍的敏感性。

圖1:CRISPR-Cas9文庫篩選確定DOCK1為二甲雙胍敏感性的決定因素
(2) 抑制DOCK1使肝癌細胞在體內和體外對二甲雙胍敏感
為了進一步描述DOCK1在臨床前模型中確定二甲雙胍敏感性中的作用,我們建立了四種患者來源的HCC類器官(即1T、2T、3T和4T)用于進一步的體外分析。進一步的免疫組化染色顯示DOCK1在所有四個類器官及其相應的腫瘤組織中表達一致(圖2A)。免疫組化染色和Western blot均顯示類器官1T和2T的DOCK1表達明顯高于3T和4T(圖2A和2B)。
為了研究HCC類器官對二甲雙胍的反應,我們用增加劑量的二甲雙胍治療四個類器官。在二甲雙胍處理下,類器官3T和4T的類器官數量和大小比1T和2T減少得更多(圖2C),表明敏感性更高,3T和4T的IC50值更低(圖2D)證實了這一點。為了研究DOCK1表達是否確實決定了患者來源的類器官中二甲雙胍的敏感性,我們在類器官1T和2T中通過shRNAs敲除DOCK1。與我們在PLC細胞中的觀察結果一致,DOCK1敲除在這些患者來源的HCC類器官中誘導二甲雙胍敏感性(圖2E-G)。此外,Ki67免疫熒光染色顯示,在DOCK1敲低的情況下,二甲雙胍能強烈抑制HCC類器官2T的增殖(補充圖未展示)。綜上所述,DOCK1表達水平有助于確定患者來源的HCC類器官的二甲雙胍敏感性。
為了研究這些體外研究結果是否可以在體內重現,我們采用了YAP5SA誘導的HCC模型。為建立該模型,采用水動力注射法將單轉座子YAP5SA和shDOCK1(或非靶向對照,NTC)表達質粒注入小鼠體內。在一個月的生長后,小劑量的二甲雙胍(100mg/kg)每天口服給這些小鼠三個月。在NTC組中,低劑量二甲雙胍對腫瘤生長沒有影響,而在shDOCK1組中,該劑量二甲雙胍可顯著降低肝癌發病率,抑制腫瘤生長(圖2H)。與這些結果一致,Ki67免疫組化染色顯示二甲雙胍治療顯著抑制了表達shDOCK1的HCC腫瘤的增殖(補充圖未展示)。Western blot分析證實YAP1在腫瘤組織中過表達,DOCK1表達下調(圖2I)。此外,使用DOCK1敲低的PLC細胞進行的小鼠異種移植實驗表明,抑制DOCK1會增強二甲雙胍的抗腫瘤作用(圖2J、2K)。總的來說,DOCK1水平調節二甲雙胍對肝癌的抗腫瘤作用的強度,而在體外和體內小鼠模型中,抑制DOCK1可使肝癌對二甲雙胍敏感。

圖2:抑制DOCK1使肝癌細胞在體內和體外對二甲雙胍敏感
(3) RAC1激活導致DOCK1介導的癌細胞對二甲雙胍不敏感
為了探索DOCK1缺乏如何增強二甲雙胍的抗腫瘤作用,我們對表達NTC或shDOCK1的PLC細胞(PLC-NTC, PLC-shDOCK1細胞)在二甲雙胍存在或不存在的情況下進行RNA-seq。在NTC細胞中,二甲雙胍改變了935個基因的表達,其中一些基因進一步受到DOCK1抑制的影響(圖3A)。為了全面解釋DOCK1在二甲雙胍介導的癌癥抑制中的作用,我們分析了三組基因,包括NTC細胞中二甲雙胍上調的基因,以及相對于二甲雙胍處理的NTC細胞,在存在或不存在二甲雙胍時,shDOCK1下調的基因。這三組基因在功能上有大量的重疊(圖3B)。進一步的基因本體(GO)和通路富集分析揭示了所有三組共有的18個GO術語或通路(圖3C)。在18個GO術語中,有4個術語與小的GTPase活性有關(圖3D),這表明小的GTPase活性通路可能參與了DOCK1抑制誘導的癌細胞對二甲雙胍的增敏。
我們重點研究了RAC家族小GTPase信號轉導通路。RAC與GTP結合時是活躍的,與GDP結合時是不活躍的。敲除DOCK1顯著降低了RAC1-GTP的水平。RAC1在細胞骨架組裝、腫瘤發生和腫瘤增殖中發揮重要作用,因此我們假設DOCK1缺乏通過抑制RAC1激活使癌細胞對二甲雙胍敏感。
Western blot顯示,二甲雙胍處理可導致PLC細胞中RAC1的激活,可見二甲雙胍存在時RAC1-GTP水平升高(圖3E)。在DOCK1敲低細胞中,二甲雙胍介導的RAC1激活被消除,這表明DOCK1是二甲雙胍激活RAC1所必需的(圖3F)。DOCK1的DOCK同源區-2(DHR2)結構域直接與無核苷酸的RAC相互作用,誘導RAC的GTP負載,從而促進其活化。因此,DHR2結構域的缺失導致DOCK1功能的喪失。為了進一步闡明shDOCK1是否通過其典型的GEF功能使癌細胞對二甲雙胍敏感,我們在內源性DOCK1敲除的PLC細胞中異位表達攜帶DHR2結構域缺失(DOCK1△DHR2)的DOCK1。
為了進一步測試RAC1激活是否對shDOCK1介導的二甲雙明敏化至關重要,我們在PLC-shDOCK1細胞中過表達野生型RAC1或RAC1G12V突變體(RAC1的組成活性形式)(圖3G)。集束形成實驗顯示,RAC1G12V的表達部分減弱了PLC-shDOCK1細胞中增強的二甲雙胍敏感性,而野生型RAC1在PLC-shDOCK1細胞中僅顯示出可忽略的影響(圖3H),這表明RAC1的激活有助于shDOCK1介導的癌細胞二甲雙胍敏化。
實時定量PCR(qPCR)和Western blot分析顯示,二甲雙胍對DOCK1的RNA或蛋白表達均無影響(圖3E)。二甲雙胍處理導致DOCK1酪氨酸殘基磷酸化增強(圖3I),這增加了DOCK1的GEF活性。為了確定哪些特定的DOCK1酪氨酸殘基在暴露于二甲雙胍時被磷酸化,我們構建了含有Y722F和Y1811F雙突變體的DOCK1Y722F/Y1811F質粒。Western blot結果顯示,與野生型DOCK1相比,二甲雙胍誘導的酪氨酸磷酸化在DOCK1Y722F/Y1811F變體中顯著降低(圖3J),這表明DOCK1Y722和Y1811殘基確實是二甲雙胍調控的磷酸化位點。
我們在PLC-shDOCK1細胞中異位表達了野生型DOCK1或DOCK1Y722F/Y1811F。Western blot結果顯示,二甲雙胍在表達野生型DOCK1的細胞中促進了RAC1的激活,但在表達DOCK1Y722F/Y1811F突變體的細胞中沒有,這表明二甲雙胍激活RAC1需要在DOCK1的Y722和Y1811殘基磷酸化(圖3K)。DOCK1Y722F/Y1811F突變體的共表達也未能減弱shDOCK1誘導的癌細胞對二甲雙胍治療的致敏性(圖3L)。綜上所述,二甲雙胍在DOCK1的磷酸化中起作用,導致RAC1的激活,DOCK1的缺乏使癌細胞對二甲雙胍敏感。

圖3:通過DOCK1磷酸化激活RAC1有助于癌細胞對二甲雙胍不敏感
(4) TBOPP與二甲雙胍在體內外的協同作用
1-(2-(30-(三氟甲基)-[1,10-聯苯]-4-基)-2-氧乙基)-5吡咯烷基磺酰-2(1H)-吡啶酮(TBOPP)是DOCK1的選擇性抑制劑。為了探索靶向DOCK1-RAC1軸的治療潛力,我們測試了TBOPP對癌細胞二甲雙胍毒性的影響。在0.75μmol/L或1μmol/L劑量下,TBOPP顯著抑制了RAC1的激活,但對PLC細胞的活力沒有影響(補充圖未展示)。然而,當1mmol/L二甲雙胍與0.75μmol/L或1μmol/L TBOPP聯合處理PLC細胞時,在降低細胞活力方面有很強的協同作用(圖4A)。Western blot檢測RAC1-GTP顯示,在PLC細胞中,TBOPP減弱了二甲雙胍誘導的RAC1激活(圖4B)。我們分別使用1.5 μmol/L和7.5 μmol/LTBOPP聯合二甲雙胍治療類器官1T和2T。結果顯示,二甲雙胍或TBOPP單獨僅輕微抑制患者來源的HCC類器官的生長和增殖,而它們的聯合治療顯著降低了兩種HCC類器官的細胞活力(圖4C),這表明二甲雙胍和TBOPP聯合使用具有強大的協同致死性。
接下來,我們使用PLC細胞進行了小鼠異種移植實驗。我們在后續實驗中選擇了8 mg/kg TBOPP劑量,以探討其與二甲雙胍的聯合作用。8 mg/kg TBOPP聯合100 mg/kg二甲雙胍可顯著抑制PLC異種移植瘤生長,且不影響小鼠體重,進一步證實了TBOPP與二甲雙胍的抗腫瘤協同作用(圖4D)。腫瘤組織裂解物的Western blot分析顯示,TBOPP治療明顯抑制二甲雙胍誘導的RAC1激活(圖4E),這證實了RAC1激活有助于體內癌細胞對二甲雙胍的DOCK1抑制相關的增敏。
為了進一步證實TBOPP和二甲雙胍之間的協同作用,我們采用NRASG12V/shP53誘導的原位肝癌模型。二甲雙胍或TBOPP單藥治療只能提供適度的腫瘤抑制,而聯合治療在NRASG12V/shP53誘導的小鼠肝癌模型中具有很強的腫瘤抑制作用(圖4F)。我們使用Ki67染色進一步證實了不同治療方法在腫瘤增殖方面的差異(圖4G),Western blot顯示TBOPP在NRASG12V/shP53誘導的原位HCC模型中消除了二甲雙胍介導的RAC1激活(圖4H)。總的來說,在體外和體內的幾個模型中,DOCK1抑制劑TBOPP與二甲雙胍在抑制肝癌方面產生了強烈的協同效應。

圖4:TBOPP與二甲雙胍在體內外的協同作用
我們通過對122例臨床HCC合并糖尿病患者的回顧性評估,試圖確定DOCK1表達水平是否可以作為評估二甲雙胍在肝癌患者治療效果的潛在生物標志物。這些患者被分為二甲雙胍使用組和其他抗糖尿病藥物使用組。我們進行免疫組化染色以可視化和定量DOCK1的表達,并根據DOCK1的平均強度將患者分為DOCK1Low(n=66)和DOCK1High (n=56)兩類。二甲雙胍似乎能顯著提高DOCK1Low患者的總生存期(圖5A),而相比之下,二甲雙胍治療與DOCK1High患者的不良預后相關(圖5B)。這些結果表明,二甲雙胍對患者的治療效果明顯相反,這取決于DOCK1水平是否相對低或高。對使用二甲雙胍的糖尿病HCC患者的進一步分析顯示,DOCK1水平低的患者總生存率更好(圖5C)。在二甲雙胍治療的糖尿病HCC患者的樣本中,Ki67染色顯示,與DOCK1High組相比,DOCK1Low組的Ki67陽性細胞比例更低(圖5D)。綜上所述,DOCK1水平決定了二甲雙胍在HCC患者中的抗腫瘤效果。
為了驗證這種可能性,我們接下來研究了DOCK1在HCC患者中的表達水平。肝癌標本中DOCK1免疫組化染色定量分析顯示,腫瘤組織中DOCK1較鄰近正常組織上調(圖5E)。qPCR和Western blot分析顯示,與匹配的相鄰非癌性肝組織相比,HCC組織中DOCK1的RNA和蛋白質水平均升高(圖5F和5G)。
DOCK1的高表達提示HCC患者二甲雙胍的療效可能較差。盡管DOCK1在HCC患者中廣泛表達上調,但腫瘤的異質性仍然導致DOCK1在HCC樣本中的差異積累,一些患者表現出非常低的,甚至無法檢測到的DOCK1水平(圖5F)。這一發現進一步表明,HCC患者應根據其特定的DOCK1水平,強烈考慮采用個性化的精準醫療方法。綜上所述,DOCK1在HCC中上調,其上調程度可決定二甲雙胍的抗腫瘤效果。

圖5:DOCK1水平決定肝癌患者中二甲雙胍的抗腫瘤活性
結論:我們的研究強調了DOCK1在決定肝癌細胞對二甲雙胍治療反應中的作用,并說明了二甲雙胍在DOCK1低表達的肝癌患者中抑制腫瘤進展。二甲雙胍-DOCK1抑制劑聯合治療DOCK1高表達肝癌患者的潛在有效性,這值得進一步的臨床研究(圖6)。
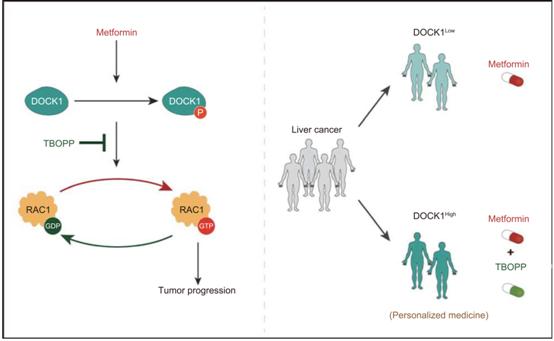
圖6:工作模型:DOCK1通過DOCK1/RAC1軸決定二甲雙胍的抗腫瘤活性。
參考文獻:
Feng, J., Lu, H., Ma, W., Tian, W., Lu, Z., Yang, H., Cai, Y., Cai, P., Sun, Y., Zhou, Z., Feng, J., Deng, J., Shu, Y., Qu, K., Jia, W., Gao, P., & Zhang, H. (2022). Genome-wide CRISPR screen identifies synthetic lethality between DOCK1 inhibition and metformin in liver cancer. Protein & cell, 13(11), 825–841. https://doi.org/10.1007/s13238-022-00906-6.