






Science高分——METRNL調控心肌修復
心肌梗死后的有效組織修復需要在不完全定義的免疫細胞-內皮細胞相互作用的指導下,產生強有力的血管生成反應,但機制不明。本研究確定單核細胞(Mos)和巨噬細胞(Mphs)衍生的細胞因子METRNL(meteorin-like)是梗死后血管生成的驅動力和干細胞因子受體KIT(KIT受體酪氨酸激酶)的高親和力配體。METRNL通過KIT依賴的信號傳導在體外培養的人類內皮細胞中介導血管生成作用。在心肌梗死的小鼠模型中,METRNL通過選擇性地擴大梗死邊緣區表達KIT的內皮細胞群來促進梗死修復。Metrnl缺失的小鼠不能產生這種KIT依賴性的血管生成反應,并在梗死后出現嚴重的心力衰竭。本研究表明,METRNL是缺血組織修復中的KIT受體配體。本文于2022年6月發表在《Science》IF:63.714期刊上。
技術路線:

主要實驗結果:
1、髓細胞來源的METRNL促進心肌梗死后血管生成
作者在急性心肌梗死小鼠模型中進行了生物信息學分泌組分析,發現了此前未鑒定到的驅動梗死修復的骨髓細胞來源的生長因子,即METRNL(meteorin-like),其在左心室梗死區域的髓系細胞中強表達(附圖1)。在假手術的基線條件下,METRNL在心臟中表達較弱,但在心肌梗死后強烈表達。共聚焦顯微鏡顯示,表達METRNL的細胞共表達髓系細胞標志物CD11b,并且定位于梗死邊緣區的內皮細胞附近(圖1A)。RT-qPCR顯示單核細胞和巨噬細胞是梗死區域,骨髓,脾臟和外周血中主要的METRNL表達的細胞(附圖2A)。Metrnl廣泛表達于梗死心臟單細胞RNA測序確定的Mo和Mph簇中(附圖2B)。根據CCR2和CX3CR1的表達來劃分Mos和Mpsh亞群,發現METRNL在CCR2high的Mos和CCR2highCX3CR1high/low的Mphs中比在CCR2lowCX3CR1high的Mphs中表達更強(附圖2C)。
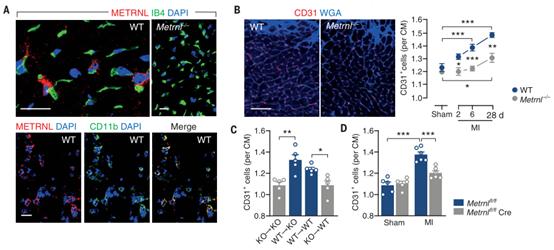
隨后用Metrnl缺陷(Metrnl-/-)小鼠建立急性心肌梗死模型,探討Metrnl在梗死修復過程中的功能。結果顯示,梗死后敲除小鼠表現出更大的梗死瘢痕,伴有更明顯的左室擴張和收縮功能障礙,并且梗死邊緣區新生毛細血管形成受損(圖1B)。在基線條件下(圖1B),Metrnl-/-小鼠的心肌毛細血管密度并未降低,這表明Metrnl缺失特異性地損害了缺血損傷后的血管生成。
用WT骨髓細胞移植Metrnl-/-小鼠可逆轉血管生成缺陷(圖1C),并改善心肌梗死后的LV瘢痕和重塑。相反,用Metrnl-/-骨髓細胞移植WT小鼠或通過Metrnlfl/fl小鼠與LysMCre/+小鼠雜交選擇性地刪除骨髓細胞中的Metrn損害了梗死后的血管生成(圖1C和D)并使LV瘢痕和重塑惡化。這些發現表明,骨髓細胞衍生的METRNL能促進血管生成、組織修復和心梗后的功能適應。
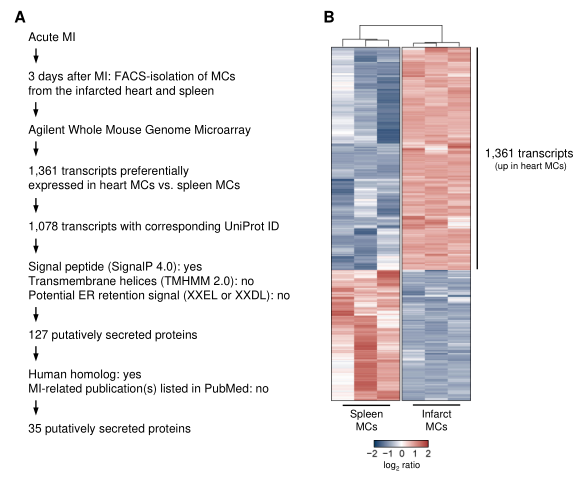
附圖1生物信息學骨髓細胞分泌組篩選
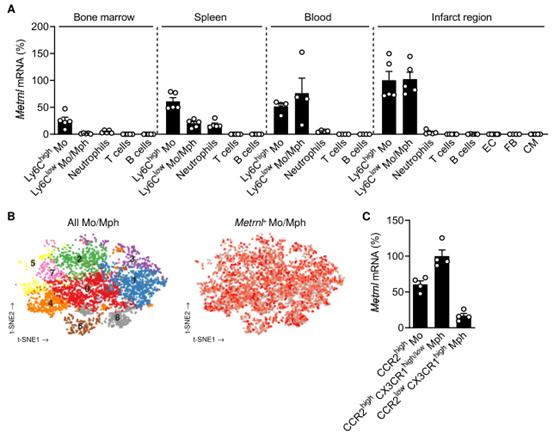
附圖2 心肌梗死后不同細胞類型Metrnl mRNA的表達
2、METRNL與KIT的細胞外結構域結合
METRNL劑量依賴性地劃痕損傷后的人冠狀動脈內皮細胞(HCAEC)增殖和遷移(圖2A-B,上),表明METRNL蛋白直接作用于ECs。使用化學交聯質譜法,確定KIT是內皮細胞中METRNL細胞表面受體的候選分子。HEK-293細胞的免疫共沉淀實驗證明了METRNL和KIT的結合。為評估METRNL是否與KIT的胞外結構域結合,從HEK-293細胞中制備表達METRNL、SCF、VEGFA,和/或分泌型KIT胞外結構域-Fc片段融合蛋白(KIT-ECD - Fc)的條件上清。從上清液中提取KIT-ECD-Fc,純化了METRNL和SCF,但沒有純化VEGFA(圖2A,下)。微尺度熱泳動顯示,METRNL和SCF以相似的高親和力結合KIT的胞外結構域(圖2B,下)。
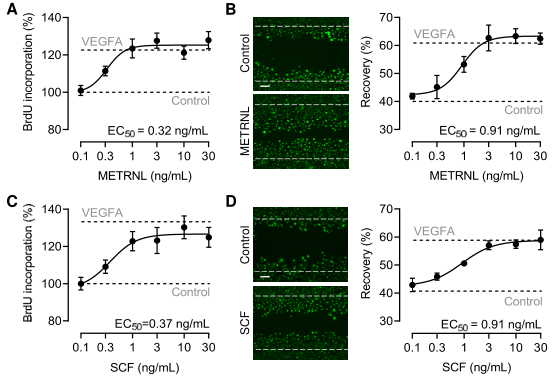

圖2 METRNL與KIT的細胞外結構域結合
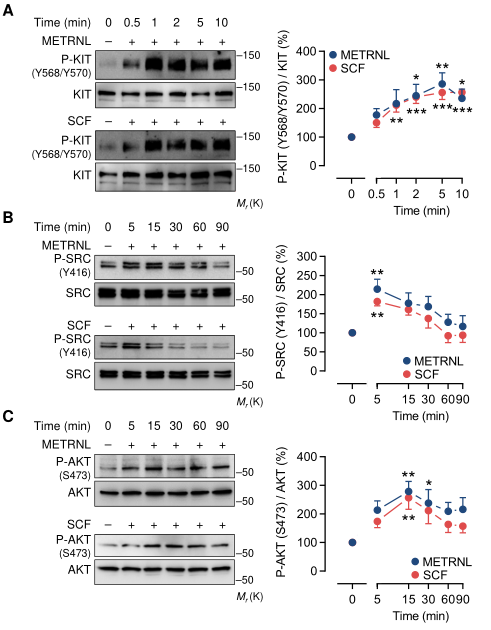
3、上皮細胞中METRNL和SCF信號通路通過KIT
SCF刺激可誘導受體近膜區酪氨酸殘基568和570 (Y568/Y570)的KIT同二聚體化和轉磷酸化,從而為SRC家族激酶創造一個對接位點。用METRNL或SCF誘導的KIT (Y568/Y570)磷酸化以相似的動力學處理HCAECs(圖3A,上)。在培養基中加入重組KIT-ECD-Fc可阻止METRNL和SCF誘導KIT (Y568/Y570)磷酸化(圖3A,下),這與跨膜KIT和KIT- ECD - Fc競爭與METRNL和SCF結合的觀點一致。事實上,重組KIT-ECD-Fc劑量依賴性地抑制了METRNL和SCF的血管生成作用,但對VEGFA的抑制作用不明顯(圖3B,下)。
KIT下游,METRNL和SCF都誘導SRC(Y416)和AKT1(S473)磷酸化,同樣遵循相似的信號動力學軌跡(圖3B-C,上)。為了獲得更廣闊的視角,應用高分辨率質譜檢測了METRNL、SCF或VEGFA刺激的HCAECs中的蛋白磷酸化動力學。在基于分布于1891個不同蛋白質的4750個磷酸化位點的主成分分析中,METRNL和SCF的磷酸化蛋白質組標簽聚在一起,并在主成分空間中與VEGFA的磷酸化蛋白質組標簽分離(圖3C,下)。因此,METRNL和SCF的血管生成作用是通過KIT和共享的下游信號通路介導的。

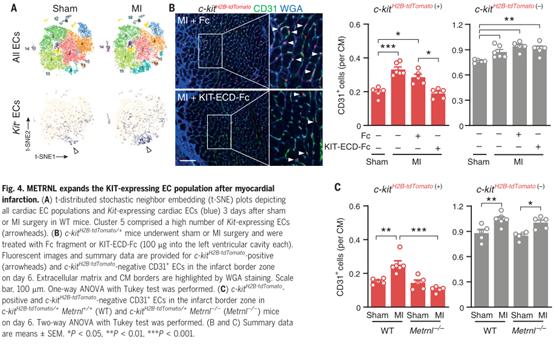
4、心肌梗死后,METRNL增加表達KIT的上皮細胞群
使用單細胞RNA測序在成年小鼠心肌中鑒定表達kit的內皮細胞。雖然在基礎條件下,少數分散在幾個EC簇的內皮細胞表達Kit,但在MI后,表達Kit的內皮細胞數量顯著增加(圖4A)。為探討KIT表達是否會影響EC對METRNL和SCF刺激的敏感性,從c-kit H2B-tdTomato/+小鼠分離心臟內皮細胞。這些小鼠的細胞核tdTomato信號完全重現了內源性KIT的表達模式。為調查KIT在梗死邊緣區的表達EC行為,用c-kit H2B-tdTomato/+小鼠構建急性心肌梗死模型。正如單細胞RNA測序觀察到的,在基線條件下和心肌梗死后,c-kit H2B-tdTomato陰性的內皮細胞數量超過c-kit H2B-tdTomato陽性的內皮細胞(圖4B)。然而,兩個人群的擴張對梗死邊緣區的毛細血管密度增加的貢獻大致相同(圖4B)。
與METRNL不同,SCF由梗死區域的ECs、成纖維細胞和心肌細胞表達(附圖3A),并且MI后SCF的表達沒有增加(附圖3B)。此外,在Metrnl- / -小鼠的梗死區域未觀察到SCF上調(附圖3C)。為研究METRNL是否驅動KIT在心肌梗死后EC中的表達,將c-kit H2B-tdTomato等位基因交叉到METRNL- / -小鼠中。在基線條件下,Metrnl缺失不影響c-kit H2B-tdTomato陽性或陰性的心肌EC密度(圖4C)。這表明在發育過程中,METRNL對于建立細胞群都不是必需的。然而,心肌梗死后,Metrnl- / -小鼠的c-kit H2B-tdTomato陽性的EC數量沒有增加,而c-kit H2B-tdTomato陰性EC擴增不受影響(圖4C)。
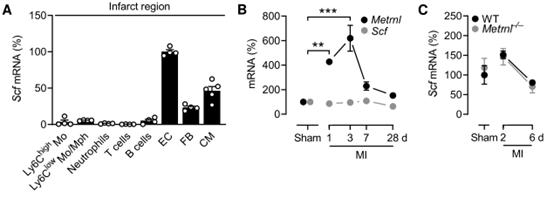
附圖3心肌梗死后SCF在心臟的表達
參考文獻:
RebollMR, Klede S, Taft MH, Cai CL, Field LJ, Lavine KJ, Koenig AL, Fleischauer J, Meyer J, Schambach A, NiessenHW, Kosanke M, van den Heuvel J, Pich A, Bauersachs J, Wu X, Zheng L, Wang Y, Korf-Klingebiel M, Polten F, Wollert KC. Meteorin-like promotes heart repair through endothelial KIT receptor tyrosine kinase. Science. 2022 Jun 17;376(6599):1343-1347. doi: 10.1126/science.abn3027. Epub 2022 Jun 16. PMID: 35709278; PMCID: PMC9838878.