






看過來!涉及鐵死亡和脂質過氧化的11分文章
鐵死亡是一種鐵依賴的細胞死亡,伴隨著脂質過氧化的積累和抗氧化系統的功能障礙。谷胱甘肽過氧化物酶4(GPX4)是肌萎縮側索硬化癥(ALS)的關鍵調節因子。然而,鐵死亡在ALS中的作用機制尚不清楚。本研究發現ALS中SPY1表達降低是由MDM2(核定位的E3泛素連接酶)介導的泛素化降解導致的。此外,SPY1通過緩解GCH1 / BH4軸(鐵死亡的一個電阻軸)失調和轉鐵受體蛋白1(TFR1)誘導的鐵產生的脂質過氧化。此外,神經元特異性過表達SPY1通過上述兩條通路顯著延緩ALS轉基因小鼠的發病并延長生存期。這些結果表明SPY1是鐵死亡和ALS的新靶點。本研究于2023年1月發表于《Cell DeathDiffer》,IF:11.84。
技術路線:

主要研究結果:
1、原代大鼠神經元轉錄組數據揭示ALS、鐵下垂和SPY1之間的關聯
為初步篩選ASL(肌萎縮性脊髓側索硬化癥)相關機制和靶基因,GEO數據庫中選取GSE7493進行分析。首先,對SOD1G93A大鼠胚胎運動神經轉錄組數據進行標準化處理,減少個體偏差的影響。接下來,使用從FerrDb下載的鐵死亡相關基因集,通過GSEA工具對所有16324個基因進行富集。ALS轉錄組數據在鐵死亡中顯著富集(圖1B)。為鑒定靶基因,進行基因差異表達分析。共篩選出417個DEGs(DEGs)。其中,SPY1被證實在小鼠和細胞中下調(圖1C)。隨后,為鑒定SPY1(Speedy/RINGO細胞周期調節器家族成員A)參與ALS的潛在效應基因,對轉錄組數據進行分組,SPY1高、低表達組。篩選出215個上調基因和205個下調基因(圖1D)。通過對比MU vsWT、SPY1高vs低和鐵死亡相關的基因集,作者發現GTP環化水解酶GCH1是唯一與ALS、鐵死亡和SPY1連鎖的基因(圖1E)。此外,David還從生物學過程、細胞組成和分子功能等方面對SPY1高、低序列中的DEGs進行功能標注表明GTPase活性也與SPY1表達相關(圖1F)。最后,通過Pearson相關性分析確定SPY1和GCH1之間的直接正相關性(圖1G)。這些生物信息學結果提示SPY1可能通過GCH1在ALS中發揮影響鐵死亡的作用。
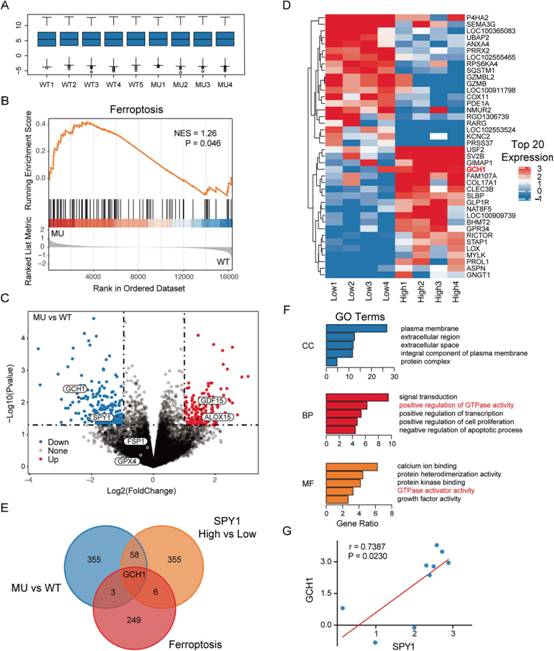
圖1原代大鼠神經元轉錄組數據揭示ALS、鐵下垂和SPY1之間的關聯
為進一步驗證鐵死亡在ALS中存在,選擇NSC34細胞作為MNs(運動神經元)體外培養的體外細胞系,驗證生物信息學分析結果。通過穩定轉染hSOD1G93A成功建立ALS模型。為初步判斷細胞死亡情況,采用CCK8發現2μM RSL3(抑制GPX4的鐵死亡誘導劑)孵育NSC34細胞2 h后,DFO和Fer-1(經典的鐵死亡抑制劑)可特異性調控NSC34細胞中的鐵死亡,而Zvad(caspase凋亡抑制劑)和Nec-1(RIPK1的壞死性凋亡抑制劑)對NSC34細胞中的鐵死亡無調控作用(圖2A)。Zvad和Nec-1對H2O2誘導的細胞凋亡具有保護作用,而鐵死亡抑制劑Fer-1沒有作用(圖2B)。然而,凋亡和鐵死亡的抑制劑均能增強hSOD1G93A細胞的活力(圖2C)。此外,這兩種抑制劑的組合顯示出明顯高于任何其他組合或任何單獨試劑的活性(圖2C)。這些結果表明細胞凋亡和鐵死亡對ALS都有貢獻。
為進行系統的研究,對鐵死亡參與的鐵積累、脂質過氧化和GSH(谷胱甘肽)消耗等典型特征進行測試。首先,通過C11-BODIPY熒光探針檢測到hSOD1G93A與WT相比發生明顯的脂質過氧化,并且可以被鐵死亡抑制劑所限制(圖2D-E)。電鏡照片顯示,hSOD1G93A引起的線粒體減少、膜密度增加和嵴破壞與鐵死亡誘導的相同(圖2F)。此外,Calcein AM試驗證實LIP升高(圖2G)。用試劑盒檢測,與對照組相比,hSOD1G93A也出現GSH的異常降低(圖2H)。qRT-PCR和蛋白質免疫印跡雜交驗證篩選的鐵死亡DEGs和關鍵基因的表達情況。突變體導致ALOX15和GDF15(分別是鐵死亡的一個驅動和一個標記)上調,GCH1和GPX4下調,FSP1未發生變化(圖1C, 2I-K)。這些結果系統地證明鐵死亡參與ALS及相關特征。
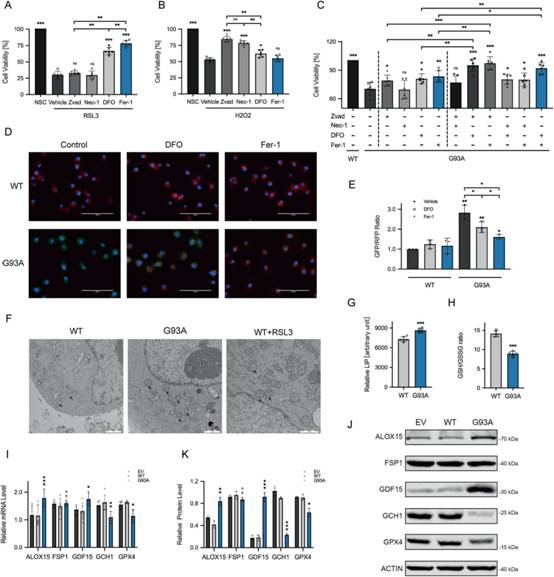
圖2鐵死亡參與ALS的細胞模型
3、MDM2在ALS中通過泛素化降解SPY1
為探究ALS中SPY1減少的原因,qRT-PCR和蛋白質免疫印跡雜交檢測轉錄和翻譯水平,發現mRNA比例下降不如蛋白明顯(圖3A-C)。在進一步的實驗中,使用MG132(蛋白酶體抑制劑)而不是胃酶抑素A與E64D(PE,自噬-溶酶體抑制劑)共孵育可以上調SPY1的表達(圖3D-E)。與預期一致,SPY1的泛素化水平在hSOD1G93A中顯著升高(圖3F)。然而,免疫共沉淀(CoIP)檢測到SPY1與SKP2或NEDD4的相互作用沒有明顯增加(圖3G)。為尋找ALS中介導SPY1泛素化的真正蛋白,利用氨基酸序列通過UbiBrowser預測具有相應組合結構的E3連接酶,其中MDM2(核定位的E3泛素連接酶)獲得最高的預測評分(圖3H)。值得注意的是,轉染MDM2后,SPY1及其泛素化水平均呈劑量依賴性增加,敲低實驗中SPY1的降解受到抑制(圖3I-K)。更令人興奮的是,CoIP證實hSOD1G93A中SPY1與MDM2的外源性和內源性相互作用比野生型增強(圖3L)。此外,在NEDD4和SKP2不變的情況下,hSOD1G93A中MDM2的表達顯著增加(圖3M-N)。這些結果闡明升高的MDM2介導SPY1的泛素化,而SPY1的泛素化主要導致ALS表達降低。
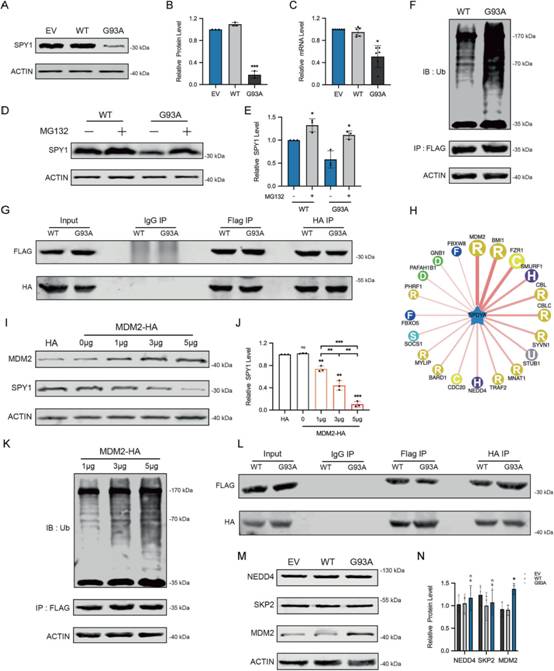
圖3ALS中SPY1的減少是由MDM2介導的
4、ALS中SPY1過表達抑制鐵死亡
為確定SPY1與鐵死亡之間的關系,在hSOD1G93A和NSC34細胞中分別構建了SPY1的過表達和敲低(圖4A)。在NSC34細胞中驗證過表達SPY1增強對RSL3誘導的鐵死亡的抗性,同時也減少hSOD1G93A細胞的死亡(圖4B-C)。在hSOD1G93A細胞中轉染SPY1后,脂質過氧化和游離鐵呈劑量依賴性減少(圖4D-F)。盡管GSH增加,但與轉染劑量無關(圖4G)。過表達SPY1對線粒體功能的保護是通過降低線粒體脂質過氧化的熒光和緩解線粒體膜電位(MMP)的下降來實現的(圖4H-I)。這些結果表明SPY1是ALS中鐵死亡的有效抑制劑。此外,其對RSL3誘導和Erastin誘導的鐵死亡的抵抗也發生在其他腫瘤細胞中(圖4J-L)。上述結果表明ALS中SPY1過表達抑制鐵死亡。

圖4過表達的SPY1在ALS細胞模型中抵抗鐵下垂
5、SPY1通過調控GCH1/BH4和TFR1抵制鐵死亡
為進一步探究SPY1調控鐵死亡的機制,首先在過表達SPY1和對照的細胞中篩選ALS中發生改變的關鍵基因的mRNA。在SPY1高vs低,上調的鐵死亡抑制因子GCH1被推測負責抵抗鐵死亡(圖1D-G)。隨后,通過過表達SPY1轉染(圖5A-B),證實GCH1的翻譯水平呈劑量依賴性上調。GCH1是BH4合成的限速酶,過表達SPY1后BH4合成顯著增加,敲低GCH1后BH4合成減少(圖5C)。同樣,敲低GCH1后,過表達SPY1引起的細胞活力和脂質氧化的變化被逆轉(圖5D-F)。然而,LIP與GCH1的表達無關(圖5G)。雖然給予BH4和過表達GCH1保護了SPY1敲低細胞的大部分活力和脂質過氧化(圖5H-I)。已知游離鐵在產生脂質過氧化中起著重要的作用,DFO和BH4的結合是偶然的,并意外地顯示出額外的改進(圖5H-I)。
為尋求不依賴于GCH1 / BH4軸對活性鐵的調控,在過表達SPY1的細胞中檢測鐵轉運相關基因的轉錄水平。與對照相比,負責轉運鐵進入細胞的TFR1(轉鐵受體蛋白1)的mRNA增加,DMT1和FPN1不變,分別負責細胞內鐵的釋放和輸出(圖5J)。與mRNA一致,蛋白水平顯示過表達SPY1降低hSOD1G93A中異常升高的TFR1(圖5K)。
為確定SPY1對鐵死亡的抵抗是否通過調節P53介導,作者檢測在SPY1-Flag細胞中過表達P53對細胞活力和相關蛋白的影響。研究發現,過表達P53引起的反向細胞死亡可以被凋亡抑制劑和鐵死亡抑制劑挽救,尤其是DFO,暗示P53與游離鐵的轉運存在聯系(圖5L)。并且隨著p53的過表達,TFR1而不是GCH1的表達上調(圖5M)。在表達SP1(S59A)突變后,作者發現SPY1可以通過上調SPY1的磷酸化水平激活GCH1(圖5N)。綜上所述,SPY1對鐵下垂的抑制主要是通過調節GCH1/BH4軸抑制脂質過氧化TFR1表達式。

圖5SPY1通過調控GCH1/BH4和TFR1抑制鐵死亡
為證明SPY1過表達對體內ALS進展的影響,將神經元特異性表達的重組病毒注射到hSOD1G93A小鼠的側腦室(圖6A)。進行性肌無力和肌萎縮是ALS的主要表現。為評估肌肉狀態,在病程中監測轉棒試驗和體重。SPY1過表達的hSOD1G93A小鼠運動能力的下降被延緩(圖6B)。體重下降的發生也延遲了約2周(圖6C)。肌無力程度和肌肉萎縮程度是影響ALS發病和生存的主導因素。與對照相比,過表達SPY1延緩了(128.90 ± 8.17 vs.120.40 ± 7.28 d)的發生,延長了(149.90 ± 12.72 vs.136.80 ± 7.98 d)的存活(圖6D-E)。
接下來,對150 d的小鼠腓腸肌和腰髓組織進行組織化學染色,以闡明SPY1的保護機制。結果顯示,SPY1顯著延緩轉棒實驗對應的hSOD1G93A小鼠神經源性萎縮(圖6F)。作為MNs的特異性標志物,脊髓前角SMI32染色發現SPY1減少了MNs的丟失(圖6F-G)。TFR1自身表達量的增加是鐵積累的有力證據,在hSOD1G93A中發現并被SPY1校正(圖6F-H)。此外,脂質氧化標志物4-HNE的免疫組化結果顯示,過表達SPY1的小鼠,其最初升高的脂質過氧化水平降低(圖6F-I)。此外,SPY1維持了較為健康的線粒體形態(圖6J)。通過免疫組化驗證TFR1的表達,發現GCH1在hSOD1G93A小鼠中表達下調,過表達SPY1后GCH1被激活(圖6K-L)。這些在hSOD1G93A小鼠中得到的結果與體外一致,強調SPY1通過抑制ALS中神經元鐵死亡來延長存活。
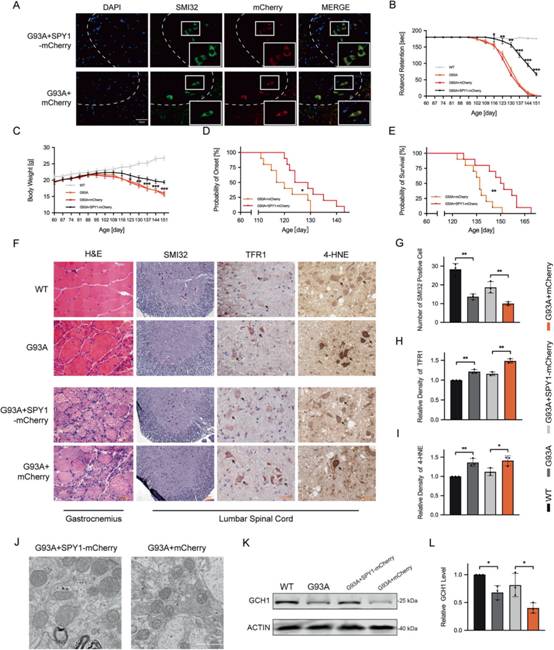
圖6SPY1在hSOD1G93A小鼠中通過抵抗神經元鐵死亡發揮保護作用。
結論:
作者通過對ALS中基因SPY1的功能富集,發現其具有轉錄正調控作用,也為SPY1通過磷酸化SP1調控GCH1的表達奠定基礎。此外,抑制或敲低USP7增強p53的泛素化,伴隨著p53水平的降低,導致TFR1的下調。作者的結果也表明SPY1通過抑制P53來調控TFR1。
參考文獻:
Wang Y, Wang C. (2023). Quantitative reactive cysteinome profiling reveals a functional link between ferroptosis and proteasome-mediated degradation. Cell DeathDiffer. 30(1):125-136. doi: 10.1038/s41418-022-01050-8.