






PKM2的乳酸化抑制促炎巨噬細胞的炎癥代謝適應
代謝適應是巨噬細胞表型轉化的重要標志和前提。丙酮酸激酶M2 (PKM2)是促炎巨噬細胞代謝適應的重要決定因素。翻譯后修飾在PKM2的調控中起核心作用。有研究首次報道了乳酸通過激活PKM2抑制Warburg效應,促進促炎巨噬細胞向修復表型的轉變,該研究報告了PKM2的一種新的翻譯后修飾類型,并闡明了其在調節促炎巨噬細胞的炎癥代謝適應中的潛在作用。發表在《INTERNATIONAL JOURNAL OF BIOLOGICAL SCIENCES》,IF:10.75。
技術路線:

主要研究結果:
1.PKM2調節LPS誘導的巨噬細胞的糖酵解
免疫細胞優先表達兩種丙酮酸激酶異構體PKM1和PKM2,數據顯示,na?ve BMDM細胞中PKM2 mRNA的表達明顯高于PKM1的表達(圖1A)。100 ng/ml LPS孵育24小時后,PKM2的mRNA表達和蛋白水平顯著增加,也顯著高于25 ng/ml IL-4誘導的巨噬細胞(圖1B, C)。雖然LPS誘導的巨噬細胞的PKM2蛋白水平顯著高于na?ve和IL-4誘導的巨噬細胞,但丙酮酸激酶活性并沒有顯著差異(圖1D)。BMDM細胞裂解物的DSS交聯表明,LPS誘導的巨噬細胞中四聚體形式的PKM2 (240KD)水平顯著低于na?ve和IL-4誘導的巨噬細胞(圖1E)。與PKM2的四聚體形式不同,二聚體/單體形式可以進入細胞核,因此我們評估了PKM2的亞細胞定位。如圖1F所示,LPS導致細胞核中PKM2的表達增加。通過免疫熒光,我們觀察到LPS誘導的巨噬細胞胞核內PKM2的水平明顯高于na?ve和IL -4誘導的巨噬細胞(圖1G)。這些結果表明PKM2在LPS誘導的巨噬細胞中過表達,但比在na?ve和IL -4誘導的巨噬細胞中更無活性。結果表明,PKM2酶抑制劑(Pi;1.2μM Compound 3K)引起LPS孵育的巨噬細胞中糖酵解速率的顯著增加(圖1H, I),但未導致OCR的變化(圖1J)。基礎OCR: ECAR比值表明,在LPS孵育的巨噬細胞中,降低PKM2丙酮酸激酶活性誘導代謝轉變為較高的糖酵解(圖1K)。這些結果表明,降低PKM2丙酮酸激酶活性增強了Warburg效應,促進了LPS孵育的巨噬細胞的炎癥代謝適應。

圖1PKM2調控脂多糖誘導巨噬細胞的糖酵解
2. 乳酸在LPS誘導的巨噬細胞中激活PKM2
PKM2調控糖酵解和乳酸生成;然而,乳酸是否調節PKM2仍不清楚。因此,我們首先檢測了乳酸對BMDM細胞中PKM2 mRNA表達和蛋白水平的影響。結果表明,外源性乳酸(20mM)的干預既沒有改變LPS誘導的巨噬細胞中PKM2的mRNA表達(圖2A),也沒有改變PKM2的蛋白水平(圖2B)。乳酸顯著促進LPS誘導的巨噬細胞的丙酮酸激酶活性(圖2C)。BMDM細胞裂解物的DSS交聯表明,LPS-LA組的PKM2四聚體形式(240KD)水平顯著高于LPS組(圖2D)。免疫熒光結果顯示,與LPS組相比,LPS-LA組細胞核中PKM2的表達明顯降低(圖2E, F)。這些結果表明,在LPS誘導的巨噬細胞中,乳酸激活了PKM2。
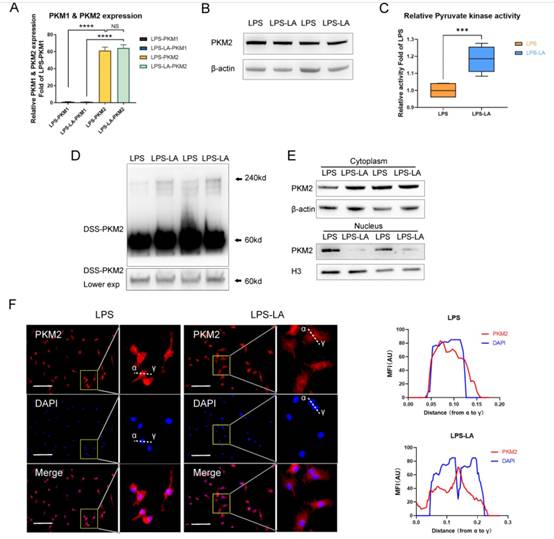
圖2乳酸激活脂多糖誘導的巨噬細胞中的PKM2
3.乳酸抑制糖酵解,并通過激活PKM2促進LPS誘導的巨噬細胞向修復表型的轉變
接下來,我們檢測了乳酸對LPS誘導的巨噬細胞ECAR和OCR水平的影響。數據顯示,乳酸顯著降低了LPS孵育的巨噬細胞中的ECAR水平(圖3A, B;LPS+LA組VS LPS組)。此外,我們觀察到乳酸降低了OCR水平(圖3C, D)。總體上,乳酸導致巨噬細胞的OCR: ECAR比值顯著上調(圖3E)。相比之下,PKM2酶抑制劑顯著增加了ECAR水平(圖3A, B;LPS+Pi組vs LPS組),但沒有改變OCR水平(圖3C, D),從而降低了OCR: ECAR比值(圖3E)。PKM2酶抑制劑顯著逆轉了乳酸的作用。與LPS+LA組相比,LPS+LA+Pi組ECAR水平較高(圖3A, B),OCR: ECAR比值較低(圖3E)。這些結果表明,乳酸可能通過激活PKM2來損害LPS誘導的巨噬細胞的炎癥代謝適應。我們分析了乳酸是否通過激活PKM2促進LPS誘導的巨噬細胞向修復表型的轉變。在BMDM細胞中,在LPS (100 ng/ml LPS)孵育48小時后,乳酸增加了ARG1水平,但降低了iNOS水平(圖3F-H;LPS-LA組vs LPS-48組)。LPS-LA-pi組iNOS水平高于LPS-LA組,ARG1水平低于LPS-LA組(圖3F-H)。這些結果支持乳酸通過激活PKM2部分促進LPS誘導的巨噬細胞向修復表型轉化的理論。
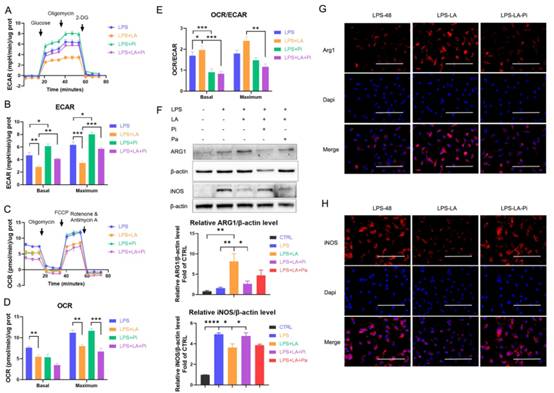
圖3乳酸抑制糖酵解并通過激活促進脂多糖誘導的巨噬細胞向修復表型的轉變PKM2
4.乳酸促進傷口巨噬細胞向修復表型的轉變,并通過激活PKM2加速小鼠傷口愈合
在本研究中,我們使用小鼠構建了皮膚傷口模型,以測試乳酸是否通過激活PKM2促進傷口巨噬細胞從促炎表型向修復表型的轉變,并加速傷口愈合。傷后第5天,LA組創面組織中促炎巨噬細胞(iNOS+ CD68+陽性細胞)數量明顯低于CTRL組,而修復性巨噬細胞(ARG1+ CD68+陽性細胞)水平高于CTRL組 (圖4A、B)。與LA組相比,LA+Pi組中促炎巨噬細胞數量較高(圖4A),而修復性巨噬細胞數量較低(圖4B)。愈合創面照片顯示,LA組小鼠創面愈合速度明顯快于CTRL組(圖4C, D),而LA+Pi組的創面愈合速度明顯慢于LA組(圖4C, D)。從數據可以看出,LA組在第9天的創面面積明顯小于CTRL組(圖4E),而第9天LA+Pi組的創面面積明顯大于LA組(圖4E)。此外,傷后第5天的再上皮化也驗證了創面愈合的速度。LA組移行上皮舌的長度明顯長于CTRL組(圖4F),而LA+Pi組的長度顯著短于LA組(圖4F)。此外,傷后第12天,三組皮膚質量無顯著差異(圖4G)。這表明乳酸干預并沒有降低傷口的愈合質量。綜上所述,這些結果表明,在小鼠中,局部給予外源性乳酸促進了傷口巨噬細胞從促炎表型向修復表型的轉變,并通過促進PKM2丙酮酸激酶活性加速了傷口愈合。
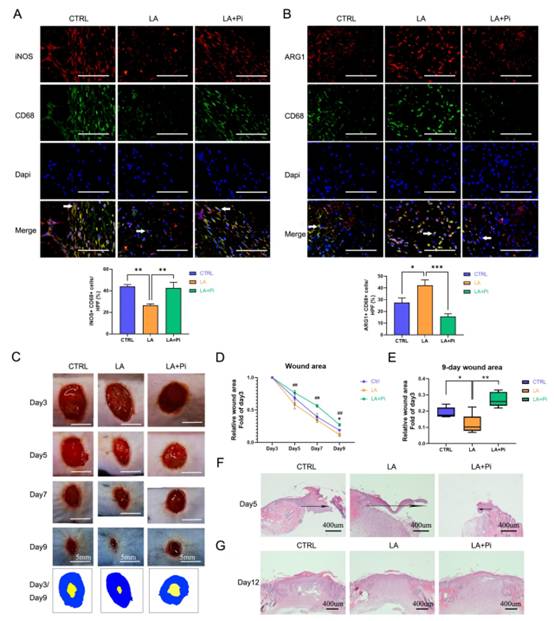
圖4乳酸通過激活PKM2促進小鼠創面巨噬細胞向修復表型轉化,加速創面愈合
5. 乳酸促進PKM2的K62乳酸化
在LPS誘導的BMDM細胞中,使用PKM2抗體下調PKM2,并使用抗lactylysine抗體檢測PKM2的乳化水平。我們觀察到PKM2被乳糖化修飾,20 mM乳酸處理24小時后,這一修飾顯著增強(圖5A)。IP質譜分析表明我們注意到4個位點(K62、K188、K224和K337)在乳酸干預后MS峰值強度增加最顯著(圖5B、C),這表明這些位點對乳酸孵養更敏感。然后,通過過表達質粒在293T細胞中異位過表達標記的PKM2,我們構建了K62R、K188R、K224R和K337R位點突變或野生型PKM2過表達293T細胞。如圖6D所示,與對照組相比,過表達PKM2的細胞中PKM2的水平明顯上調。接下來,我們使用標志抗體拉低PKM2來檢測泌乳水平。結果證實,K62R位點突變顯著降低了PKM2的泌乳水平,而其他三個位點突變對泌乳水平無顯著影響(圖5E)。這些結果表明K62位點是PKM2的主要泌乳位點。PKM2的三級結構表明,K62位于PKM2蛋白的A結構域(圖5F),并且與關鍵活性位點S362相鄰。如圖5G所示,K62在從達尼奧到各種哺乳動物的物種中都是保守的。這些事實支持K62位點是PKM2功能的潛在調控位點的假設。
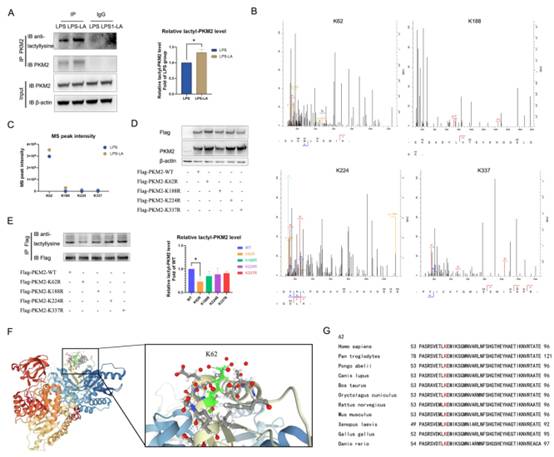
圖5乳酸促進PKM2的K62乳酸化
6.K62R突變體逆轉了乳酸對PKM2丙酮酸激酶活性和巨噬細胞表型轉變的調節
我們進一步評估了K62位點乳酸化對PKM2功能的調節作用。如圖6A所示,乳酸孵育顯著促進了WT型PKM2過表達293T細胞中PKM2酶的活性。K62R突變顯著抑制了乳酸培養后293T細胞中的PKM2酶活性(圖6B;K62R-LA組vs WT-LA組)。乳酸孵育293T細胞的裂解產物的DSS交聯表明,K62R-LA組的PKM2四聚體形式(240KD)水平顯著低于WT-LA組(圖6C)。此外,在乳酸孵育的293T細胞中,K62R-LA組細胞核中PKM2的水平約為WT-LA組的1.41倍(圖6D)。這些結果證實了K62位點對PKM2的功能有明顯的影響,并表明乳酸通過K62位點的泌乳部分促進PKM2酶的活性。接下來,我們使用慢病毒載體在BMDM細胞中過表達K62R突變或野生型PKM2。LPS孵育48小時后,乳酸增加了WT BMDM細胞中ARG1的水平,但降低了iNOS的水平(圖6E-G;WT-LPS- la組vs WT-LPS組)。乳酸的這一作用在K62R突變的BMDM細胞中被顯著削弱(圖6E-G;K62R-LPS- LA組vsK62R-LPS組)。與WT-LPS-LA組相比,K62R-LPS-LA組顯示出較高的INOS水平和較低的ARG1水平(圖6E-G)。這些結果支持以下結論:乳酸通過K62位點PKM2的乳化,部分促進了LPS誘導的巨噬細胞向修復表型的轉變。
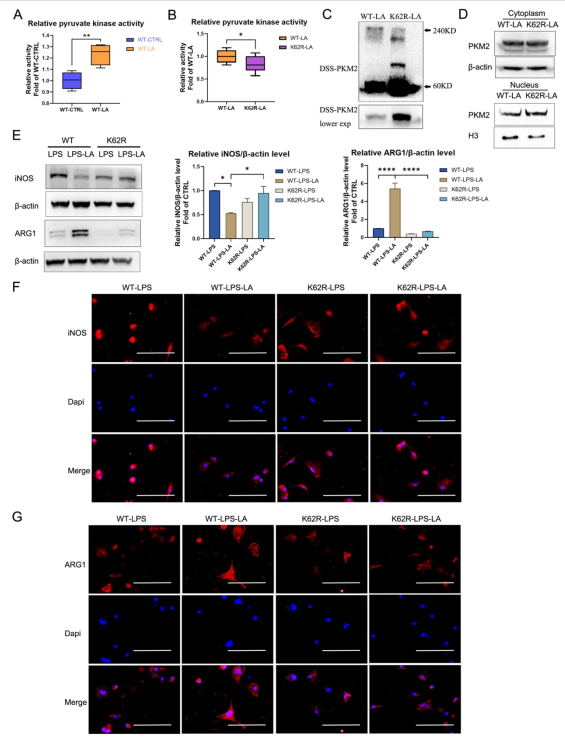
圖6K62R突變體逆轉了乳酸對PKM2丙酮酸激酶活性和巨噬細胞表型轉變的調節
結論:
綜上所述,該研究首次確定了PKM2作為一種乳化底物。乳糖化增加PKM2的丙酮酸激酶活性,減少其四聚體到二聚體的轉變和核分布。乳酸通過激活PKM2導致糖酵解減少,促進促炎巨噬細胞向修復表型的轉變。