






LncRNA DACH1通過與SRSF1結合抑制CTNNB1積累來防止肺纖維化
肺纖維化是多種肺部疾病的終末期結果,其特征是成纖維細胞過度增殖、異常細胞外基質 (ECM) 沉積伴有炎癥損傷和組織結構破壞。最常見的疾病類型是特發性肺纖維化 (IPF),這是一種嚴重的間質性肺疾病,會導致肺功能進行性喪失,由于其嚴重的臨床表現、預后不良和高死亡率導致重大醫療負擔。IPF的分子機制尚不完全清楚,除肺移植外,IPF尚缺乏有效的治療方法。該研究發表于《ACTA PHARMACEUTICA SINICA B》,IF: 14.903。
技術路線:

主要研究結果:
1. LncDACH1缺乏導致小鼠肺纖維化
在之前的研究中,作者生成了LncDACH1敲除 (LncDACH1-KO) 小鼠,其表現出肺中LncDACH1的顯著減少。出乎意料的是,根據顯微計算機斷層掃描(Micro-CT,圖1A)。此外,LncDACH1- KO小鼠的用力肺活量 (FVC)、用力呼氣流量 (FEF)、FEV0.1、FEV0.1/FVC、吸氣能力 (IC) 以及流量-容積環也有所減弱(圖1B),說明LncDACH1與小鼠肺功能密切相關。作者進一步檢測到LncDACH1的缺失會誘導Fn1、Col1a1、Col3a1和Acta2的mRNA水平,以及與肺纖維化相關的FN1和膠原蛋白I的蛋白表達(圖1C和D)。羥脯氨酸在膠原蛋白中含量最豐富,這一指標因敲除LncDACH1而升高(圖1E)。在肺組織切片上使用H&E染色和Masson三色染色,作者觀察到LncDACH1的敲除促進了膠原沉積并增加了纖維化面積(圖1F和G)。與這些結果一致,免疫組織化學和免疫熒光證實了LncDACH1-KO小鼠肺中膠原蛋白I和α-SMA的表達上調(圖1H-K),表明LncDACH1缺陷小鼠表現出自發性肺纖維化。

圖1 LncDACH1的缺失導致小鼠肺纖維化
為了評估LncDACH1在肺纖維化中的作用,作者檢測了LncDACH1在肺瘢痕灶中的表達。qRT-PCR分析顯示LncDACH1的表達在IPF患者的肺組織和BLM治療的小鼠中下調。
考慮到細胞的異質性,作者對從IPF和正常肺中分離的細胞進行了scRNA-seq分析。首先,IPF Cell Atlas ( http://ipfcellatlas.com/ ) 分析了GSE135893數據集。基于相對相似性,形成了四種細胞類型組:上皮細胞、內皮細胞、間充質細胞和免疫細胞。作者發現LncDACH1保守序列DACH1的表達在成纖維細胞中高于AT2細胞。來自人類蛋白質圖譜網站 ( https://www.proteinatlas.org/ ) 的ScRNA-seq結果顯示DACH1在內皮細胞、粒細胞和成纖維細胞中高度表達。與體內結果一致,在從BLM誘導的成年小鼠分離的肺成纖維細胞和TGF-β1誘導的原代小鼠肺成纖維細胞 (PMLF)中,LncDACH1的表達均顯著降低。
2. LncDACH1的過表達減輕了TGF-β1誘導的小鼠肺成纖維細胞纖維化
為了進一步了解LncDACH1是否作用于成纖維細胞,作者構建了LncDACH1質粒以成功增強LncDACH1在PMLF中的表達(圖2A)。TGF-β1誘導的PMLFs 中Fn1、Col1a1、Col3a1和Acta2的mRNA表達上調,但在LncDACH1過表達后這些改變被逆轉(圖2B)。同時,LncDACH1轉染的PMLFs中FN1和膠原蛋白I的蛋白表達低于TGF -β1誘導的細胞(圖2C)。由于成纖維細胞的激活是肺纖維化的一個重要特征,作者評估了LncDACH1對PMLF功能的調節作用。根據EdU熒光染色,LncDACH1被證實可抑制由TGF-β1誘導的細胞增殖增加(圖2D)。傷口愈合試驗的結果表明,LncDACH1抑制肺成纖維細胞的遷移能力(圖2E)。免疫熒光染色還顯示,在LncDACH1的存在下,成纖維細胞向α- SMA 陽性肌成纖維細胞的轉變減弱(圖2F)。

圖2 LncDACH1在原代小鼠肺成纖維細胞中的抗纖維化作用
3. LncDACH1與SRSF1結合以調節其表達和活性
lncRNA 的細胞定位很大程度上決定了它們的作用機制。因此,作者設計了FISH探針,發現LncDACH1在細胞質和細胞核中均有表達,但在生理條件下主要定位于細胞核(圖3A)。最近,有研究報道lncRNAs可以直接與蛋白質結合,然后調節蛋白質的表達和活性。作者假設LncDACH1通過與核蛋白相互作用參與纖維發生。通過使用RNA結合蛋白相互作用數據庫,包括RBPmap(http://rbpmap.technion.ac.il/manual.html)、RBBPB(http://rbpdb.ccbr.utoronto.ca/)和catRAPID(http ://service.tartaglialab.com/page/catrapid_group ) , 預測與LncDACH1結合的蛋白質(圖3B)。在9種預測的蛋白質中,SRSF1引起了作者的興趣,因為它主要與LncDACH保守序列結合。如圖3C所示,LncDACH1-KO小鼠肺中SRSF1蛋白水平升高。轉染LncDACH1過表達質粒的PMLFs抑制了TGF-β1誘導的SRSF1上調(圖3D)。此外,作者使用翻譯抑制劑放線菌酮以時間依賴性方式檢測LncDACH1對SRSF1穩定性的影響。蛋白質分析表明,LncDACH1的過表達增強了SRSF1蛋白的降解(圖3E)。這些結果表明LncDACH1不依賴蛋白酶體或泛素化來降解SRSF1。作者接下來進行了RNA下拉分析,以探索LncDACH1是否直接與SRSF1結合。正如預期的那樣,在提取 RNA-蛋白質復合物后,通過蛋白質印跡檢測SRSF1的蛋白質表達(圖3F)。進行RNA結合蛋白免疫沉淀 (RIP) 測定以評估LncDACH1和SRSF1之間的關系。與IgG沉淀相比,作者觀察到LncDACH1在抗SRSF1抗體沉淀中的富集(圖3G)。此外,過表達LncDACH1明顯減弱了PMLF中的SRSF1熒光(圖3H)。總之,作者的結果表明LncDACH1與SRSF1結合以調節其表達。

圖3 LncDACH1調節SRSF1表達
4. SRSF1是LncDACH1的抗纖維化作用所必需的
為了研究LncDACH1與SRSF1相互作用在肺纖維化中的作用,作者同時將LncDACH1過表達質粒和含有腺病毒的SRSF1過表達質粒 (Adv-SRSF1) 轉染到PMLF中。在mRNA水平上,增強的SRSF1表達抑制了LncDACH1對Fn1、Col1a1、Col3a1和Acta2的負調控(圖4A)。SRSF1對FN1和膠原蛋白I的蛋白表達也有促進作用(圖4B),表明SRSF1的表達恢復了LncDACH1對纖維化和膠原沉積的抑制作用。同樣,與過表達LncDACH1的細胞相比,SRSF1增加的PMLF 表現出更強的增殖能力(圖4C)。在TGF-β1的背景下,SRSF1阻礙了LncDACH1對細胞遷移的抑制作用,并促進了成纖維細胞向肌成纖維細胞的轉變(圖4D和E)。這些發現表明,SRSF1作為LncDACH1的下游靶基因,介導LncDACH1的抗纖維化作用。

圖4 SRSF1的過表達限制了LncDACH1的功能
5. LncDACH1通過靶向SRSF1抑制Ctnnb1表達
先前的研究表明,SRSF1通過募集Ctnnb1 mRNA并增強其生物合成來促進β-catenin 蛋白的積累。Ctnnb1是Wnt信號通路的關鍵效應子,參與肺纖維化的發病機制。作者從LncDACH1-KO小鼠的肺組織中進行蛋白質印跡分析,發現β-catenin蛋白水平顯著上調(圖5A)。隨后,作者發現LncDACH1在體外可以逆轉TGF-β1誘導的β-catenin表達增加(圖5B)。然而,LncDACH1對β-catenin蛋白的抑制被SRSF1過表達消除(圖5C)。此外,免疫熒光染色顯示,LncDACH1的強制表達抑制了TGF-β1處理的PMLFs中β-catenin的表達和核轉位,而這種作用被SRSF1過表達顯著減弱(圖5D)。

圖5 沉默Ctnnb1消除了LncDACH1缺乏的促纖維化作用
接下來,作者構建了小干擾 RNA (siRNA) 來沉默LncDACH1和Ctnnb1,并驗證了它們的效率。如圖5E所示,LncDACH1的沉默促進了β-catenin的表達和核轉位,而這種作用被Ctnnb1敲低顯著抑制。qRT-PCR結果顯示,LncDACH1缺陷導致Fn1、Col1a1、Col3a1和Acta2上調,而沉默Ctnnb1則逆轉上述變化(圖5F)。敲低Ctnnb1可抑制由LncDACH1缺陷引起的FN1、膠原蛋白 I、SRSF1和β-catenin蛋白表達增加(圖5G)。此外,敲低LncDACH1足以誘導PMLFs的增殖和遷移,而敲低Ctnnb1則消除了這種情況(圖5H和I)。免疫熒光結果顯示,在LncDACH1沉默的細胞中,α-SMA的表達增加,而Ctnnb1的抑制逆轉了成纖維細胞向肌成纖維細胞的分化(圖5J)。因此,作者的數據表明,Ctnnb1是LncDACH1靶向SRSF1調節肺纖維化的關鍵步驟。
6. LncDACH1的增強表達可預防BLM誘導的小鼠肺纖維化
為了驗證LncDACH1是否在體內發揮抗纖維化作用,作者構建了一個含有LncDACH1的腺病毒相關病毒5 (AAV-5) 過表達質粒,命名為AAV5-LncDACH1。作者在BLM誘導前14天氣管內施用AAV5-LncDACH1,以觀察LncDACH1對肺纖維化的預防作用。在BLM注射后第21天對小鼠實施安樂死,并獲得肺組織用于后續研究(圖6A)。根據qRT-PCR分析,Fn1、Col1a1、Col3a1和Acta2的 mRNA 水平被預防性LncDACH1抑制(圖6B)。蛋白質印跡檢測到BLM對FN1和膠原蛋白I的誘導受到AAV5-LncDACH1的阻礙(圖6C)。此外,LncDACH1的下游靶標SRSF1和β-catenin也同樣被下調(圖6C)。與BLM處理后 AAV5-pcDNA3.1處理的小鼠相比,AAV5- LncDACH1處理的小鼠肺組織中的羥脯氨酸含量較低(圖6D)。肺組織用多聚甲醛固定并進行H&E和Masson染色后,預先注射AAV5 -LncDACH1的小鼠肺組織形態和纖維化面積有明顯改善,說明LncDACH1在體內抑制了纖維化發生(圖6E和F)。免疫組化和免疫熒光顯示,BLM誘導的膠原蛋白I和α-SMA表達增加被LncDACH1預防顯著抑制(圖6G-J)。這些結果說明LncDACH1在BLM誘導的肺纖維化中起保護作用。

圖6 LncDACH1抑制BLM誘導的肺纖維化
7. 在已建立的小鼠肺纖維化實驗模型中,LncDACH1的強制表達減弱了肺纖維化的進展
由于肺纖維化的誘因復雜,病程長,早期認識和治療是提高生存率的有效方法。在治療方案中,作者在BLM誘導后5天注射AAV5-LncDACH1,并在BLM氣管內給藥后21天處死小鼠(圖7A)。在BLM誘導小鼠肺纖維化后,纖維化相關基因的mRNA和蛋白水平受到LncDACH1表達增強的抑制(圖7B和C)。SRSF1和β-catenin的蛋白表達同樣受到LncDACH1處理的調節(圖7C)。LncDACH1處理后羥脯氨酸含量與正常對照小鼠沒有明顯差異,但在BLM暴露的小鼠中羥脯氨酸含量顯著降低(圖7D)。響應于治療性AAV5- LncDACH1治療,小鼠肺組織顯示出較少的纖維化區域,較輕的膠原蛋白I和α-SMA 染色(圖7E-J),表明LncDACH1能夠阻止肺纖維化的進展。更重要的是,作者同時在小鼠中過表達了LncDACH1和SRSF1。作者的結果表明,LncDACH1減弱了BLM誘導的小鼠纖維化,而SRSF1逆轉了LncDACH1的功能。這些結果證明了LncDACH1 /SRSF1/ Ctnnb1軸的抗纖維化作用。

圖7 治療性LncDACH1對肺纖維化的抑制作用
8. LncDACH1保守序列在MRC-5細胞中的抗纖維化作用
LncDACH1在小鼠模型和原代小鼠肺成纖維細胞中表現出對肺纖維化的有效保護作用。作者希望確定LncDACH1是否對人源成纖維細胞(MRC-5細胞)也有相同的作用。由于LncDACH1過長的核苷酸序列限制了其藥用潛力,作者鑒定了可能與SRSF1結合的 LncDACH1序列(800-1200)(如圖3B),基于先前的一項研究,該研究表明LncDACH1 (835–1251) 片段具有功能性且高度保守。作者通過RNA下拉試驗檢測到SRSF1和LncDACH1 (835-1251) 相互作用(圖3H)。qRT-PCR分析顯示LncDACH1 (835-1251) 在MRC-5細胞中的表達在被TGF-β1誘導后被抑制(圖8A)。在纖維化方面,LncDACH1 (835-1251) 顯著減輕TGF-β1增加FN1、COL1A1、COL3A1和ACTA2的mRNA表達,同時抑制FN1、膠原蛋白 I、β-連環蛋白和SRSF1蛋白水平的上調,以及效率與全長LncDACH1一致(圖8B和C)。此外,作者發現LncDACH1 (835-1251) 抑制 MRC-5 細胞的遷移、增殖和分化(圖8D-F)。免疫熒光分析顯示LncDACH1 (835-1251)通過減少β-連環蛋白的核轉位來抑制其活性(圖8G)。總之,作者已經證明LncDACH1的保守序列對人胎肺成纖維細胞的活化具有改善作用。
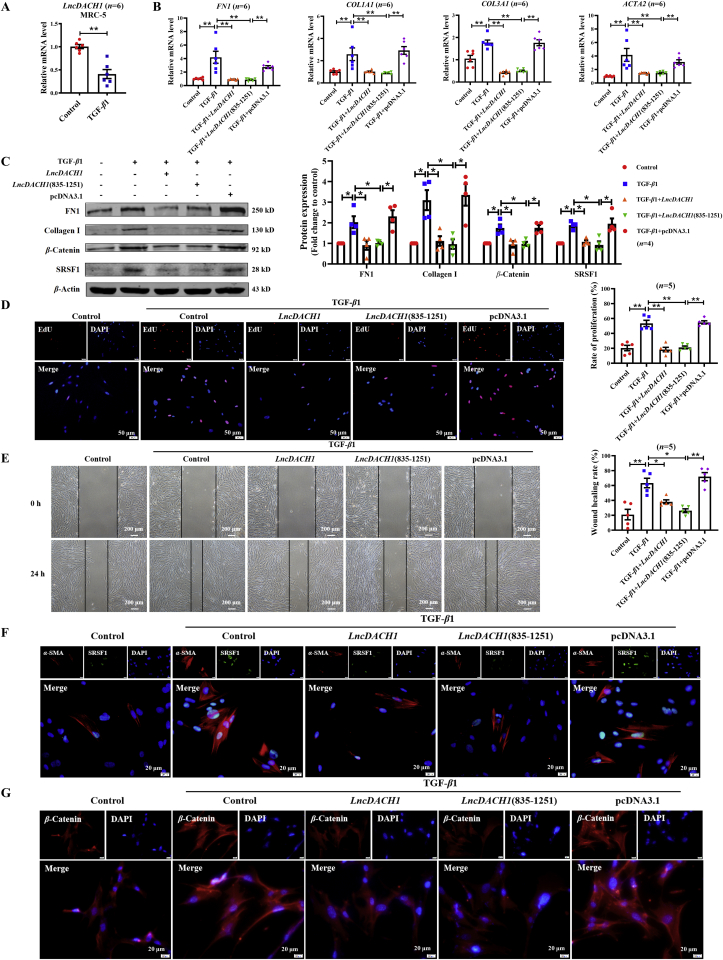
圖8 LncDACH1的保守序列對人肺成纖維細胞(MRC-5)的影響
結論:
該研究表明,LncDACH1通過與SRSF1結合并抑制SRSF1的表達來負調節CTNNB1的水平,從而抑制肺成纖維細胞的活化和細胞外基質的沉積,從而參與緩解肺纖維化的過程。因此,可以利用LncDACH1的替代來增強其表達以預防和治療IPF。
圖形摘要:
LncDACH1通過與SRSF1結合并抑制SRSF1蛋白表達負調控CTNNB1,從而抑制肺成纖維細胞的活化和細胞外基質的沉積,從而減輕肺纖維化。

參考文獻:
Sun J, Jin T, Niu Z, Guo J, Guo Y, Yang R, Wang Q, Gao H, Zhang Y, Li T, He W, Li Z, Ma W, Su W, Li L, Fan X, Shan H, Liang H. LncRNA DACH1 protects against pulmonary fibrosis by binding to SRSF1 to suppress CTNNB1 accumulation. Acta Pharm Sin B. 2022 Sep;12(9):3602-3617. doi: 10.1016/j.apsb.2022.04.006. Epub 2022 Apr 16. PMID: 36176913; PMCID: PMC9513499.