






ALKBH5可能通過降低PHF20 mRNA甲基化抑制結直腸癌進展
m6A是最廣泛的mRNA修飾,被甲基轉移酶和去甲基酶動態地、可逆地調控。ALKBH5是一種主要的去甲基化酶,在癌癥的進展中起著至關重要的作用。然而,ALKBH5在結直腸癌(CRC)中的作用和機制尚不清楚。我們發現,在CRC中,ALKBH5的下調與CRC患者預后不良密切相關。在功能上,敲低ALKBH5可增強LOVO和RKO細胞的增殖、遷移和侵襲能力,而過表達ALKBH5可抑制這些細胞的功能。結果還表明,敲除ALKBH5可促進LOVO在體內的皮下腫瘤發生,而過表達ALKBH5可抑制這種能力。在機制上,MeRIP-seq和RNA-seq的聯合分析結果表明,PHF20 mRNA是一個受ALKBH5介導的m6A修飾調控的關鍵分子。進一步的實驗表明,ALKBH5可能通過去除PHF20 mRNA 3'UTR的m6A修飾來抑制PHF20 mRNA的穩定性。總之,ALKBH5通過降低PHF20 mRNA甲基化抑制CRC進展。ALKBH5介導的PHF20 mRNA的m6A修飾有望成為CRC的干預和治療策略。本文于2022年6月發布于“Clinical and Translational Medicine”(IF=8.554)上。
技術路線

結果:
1)ALKBH5在CRC中表達下調
為了評估ALKBH5在癌癥中的表達,我們初步檢測了TCGA數據集中23個實體癌癥中ALKBH5的mRNA表達。在12例實體癌中,與相鄰正常組織相比,ALKBH5明顯失調(圖1A)。與相鄰正常組織相比,ALKBH5在7種實體癌(CRC、GBM、KICH、KIRP、PRAD、THCA和UCEC)中顯著下調,在5種實體癌(CHOL、HNSC、KIRC、LIHC和LUSC)中上調。我們發現,與相鄰正常組織相比,TCGA-CRC隊列中腫瘤組織中的ALKBH5明顯減少。我們還從GEO數據集中下載了CRC的RNA測序數據。結果證實了CRC組織中ALKBH5 mRNA的減少(圖1 B)。RT-qPCR結果顯示,CRC組織中ALKBH5 mRNA表達明顯低于相鄰正常組織(圖1C)。我們還評估了ALKBH5蛋白在CRC中的表達。免疫組化檢測結果顯示,CRC組織中ALKBH5蛋白表達明顯降低(圖1D,E)。此外,mRNA的m6A 定量分析表明,CRC組織中mRNAs的m6A修飾水平較高(圖1F)。

2)ALKBH5缺失預示CRC患者預后較差
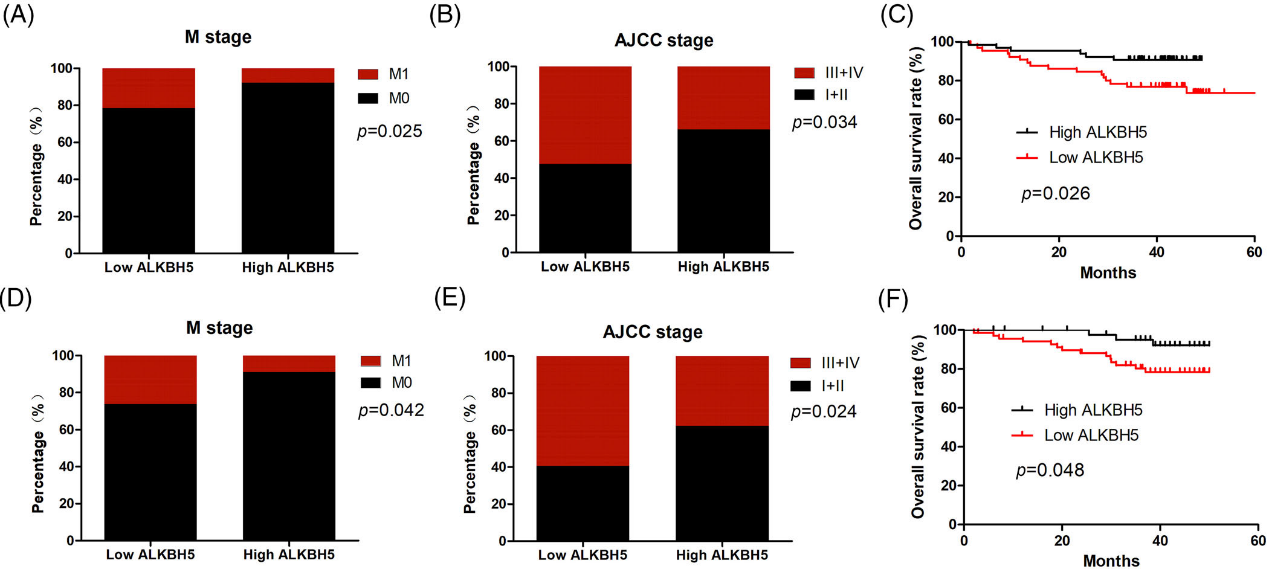
為了探討ALKBH5的臨床價值,我們從mRNA和蛋白水平分析了ALKBH5與CRC患者臨床病理特征的相關性。結果表明,在mRNA水平上,下調ALKBH5與遠處轉移和臨床晚期顯著相關(圖2A, B)。一致地,在蛋白水平上,下調的ALKBH5與遠處轉移和臨床晚期顯著相關(圖2D, E)。Kaplan-Meier分析表明,缺失ALKHB5的CRC患者在mRNA(圖2C)和蛋白(圖2F)水平上的總生存期都較短。
3)敲除ALKBH5可促進結腸癌細胞的生長和轉移
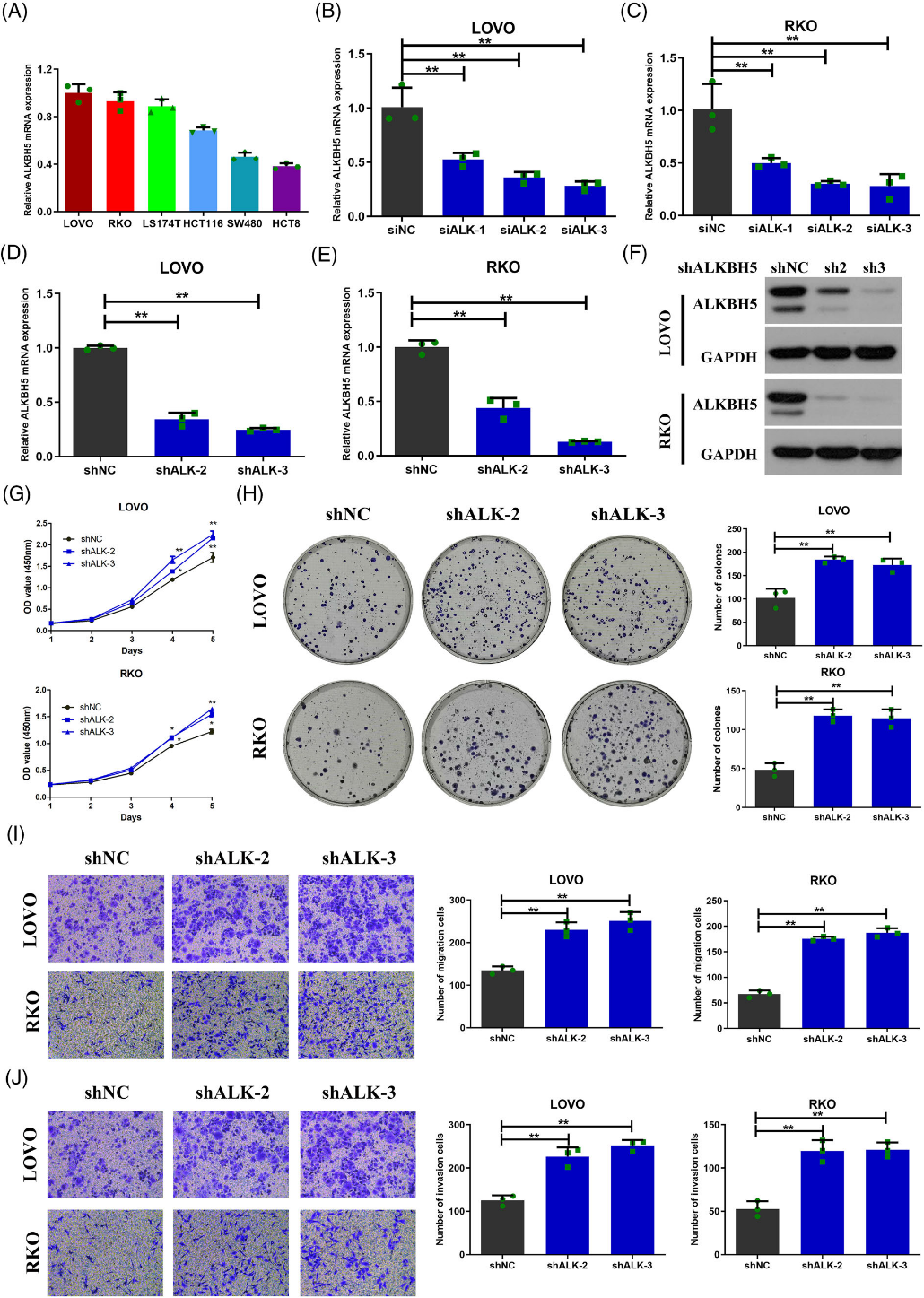
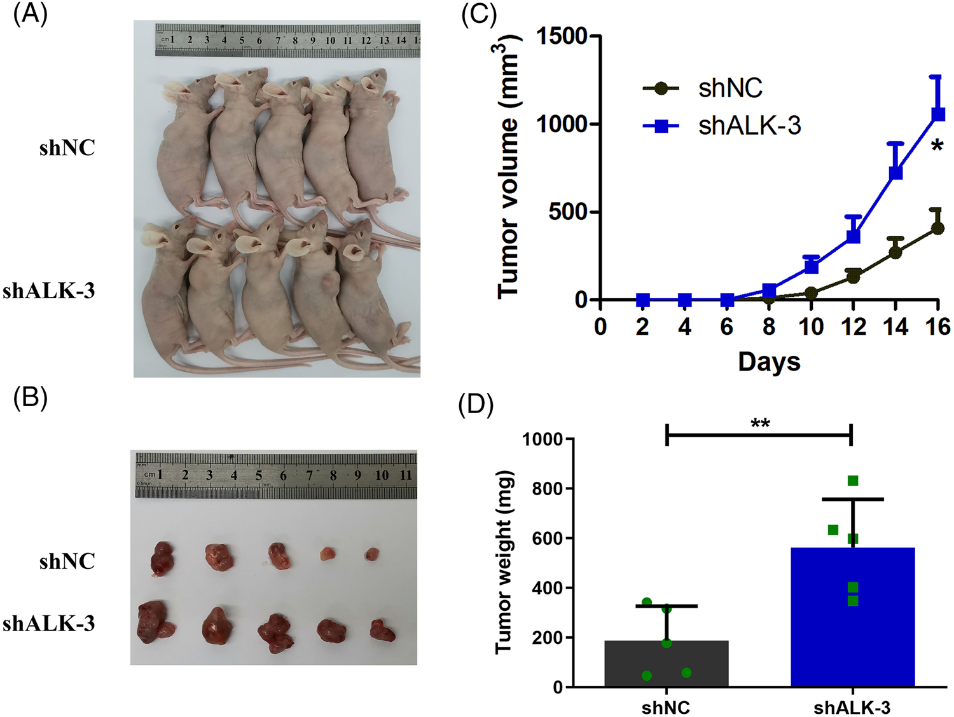
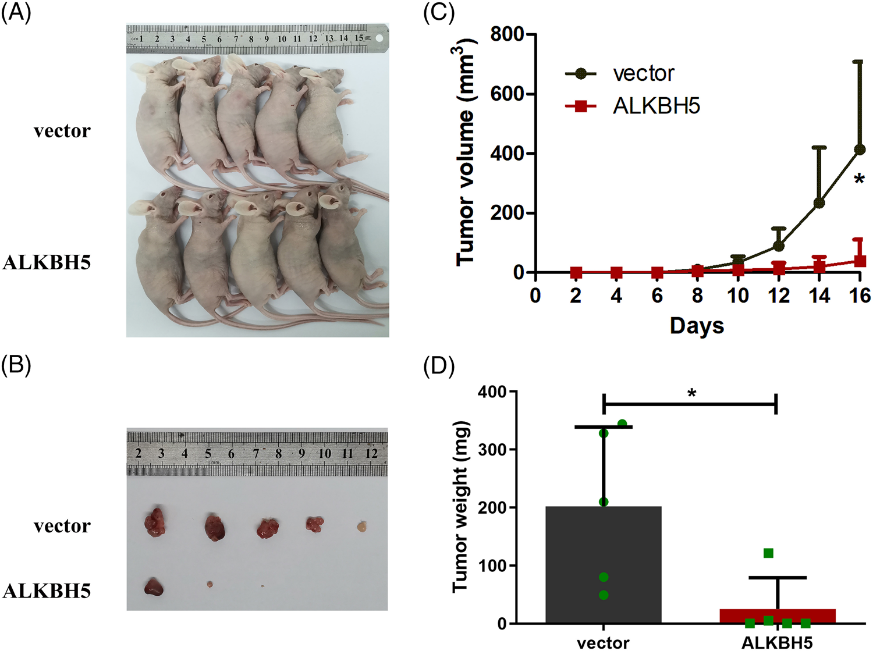
我們探索了ALKBH5對結腸癌細胞功能的影響。我們初步鑒定了五種結腸癌細胞中ALKBH5的mRNA表達,并注意到與其他結腸癌細胞系相比,LOVO和RKO細胞中ALKBH5表達升高(圖3A)。在LOVO和RKO細胞中使用三種特定的siRNA敲除ALKBH5(圖3B,C)。利用敲除效率高的兩個siRNA (siALKBH5-2, siALKBH5-3)構建敲除質粒(shALKBH5-2, shALKBH5-3),用于后續實驗。我們使用兩個獨立的ALKBH5敲除慢病毒構建了兩個穩定的細胞系。在mRNA和蛋白水平上驗證敲除效率(圖3D-F)。隨后,CCK-8實驗顯示,ALKBH5敲除顯著增強了細胞生長,以及LOVO和RKO細胞的活力(圖3G)。通過克隆形成實驗證實,敲除ALKBH5可顯著提高LOVO和RKO細胞的克隆形成能力(圖3H)。此外,我們使用transwell實驗評估了LOVO和RKO細胞的遷移和侵襲能力。結果表明,敲除ALKBH5可顯著提高細胞遷移和侵襲能力(圖3I,J)。為了進一步確定ALKBH5在體內是否影響結腸癌細胞功能,我們建立了異種移植瘤模型。我們發現,當ALKBH5敲低的LOVO細胞植入時,腫瘤生長速度更快(圖4C),腫瘤體積和重量增加(圖4A,B,D)。
4)過表達ALKBH5可抑制結腸癌細胞的生長和轉移
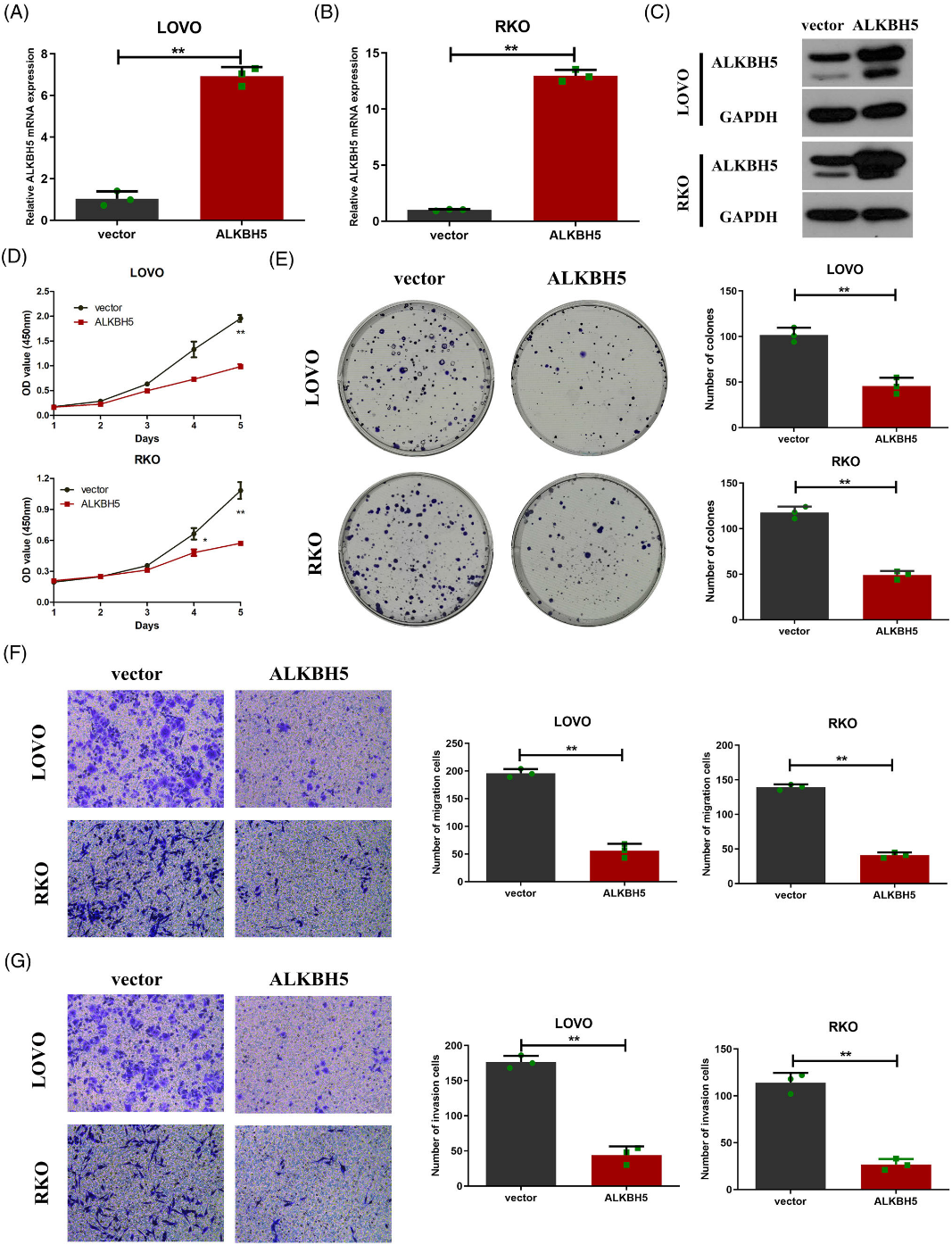
我們還利用慢病毒載體構建了過表達ALKBH5的穩定轉染細胞系,RT-qPCR和Western blot實驗驗證了過表達效率(圖5A-C)。CCK-8實驗顯示,過表達ALKBH5顯著抑制細胞生長和LOVO和RKO細胞的活力(圖5D)。集落形成實驗證實,過表達ALKBH5導致LOVO和RKO細胞集落形成能力顯著降低(圖5E)。此外,通過transwell分析,過表達ALKBH5大大削弱了細胞遷移和侵襲能力(圖5F,G)。然后,我們的腫瘤異種移植模型的結果表明,當移植過表達ALKBH5的LOVO細胞時,腫瘤生長速度較慢(圖6C),腫瘤體積和重量降低(圖6A,B,D)。
5)ALKBH5靶向PHF20
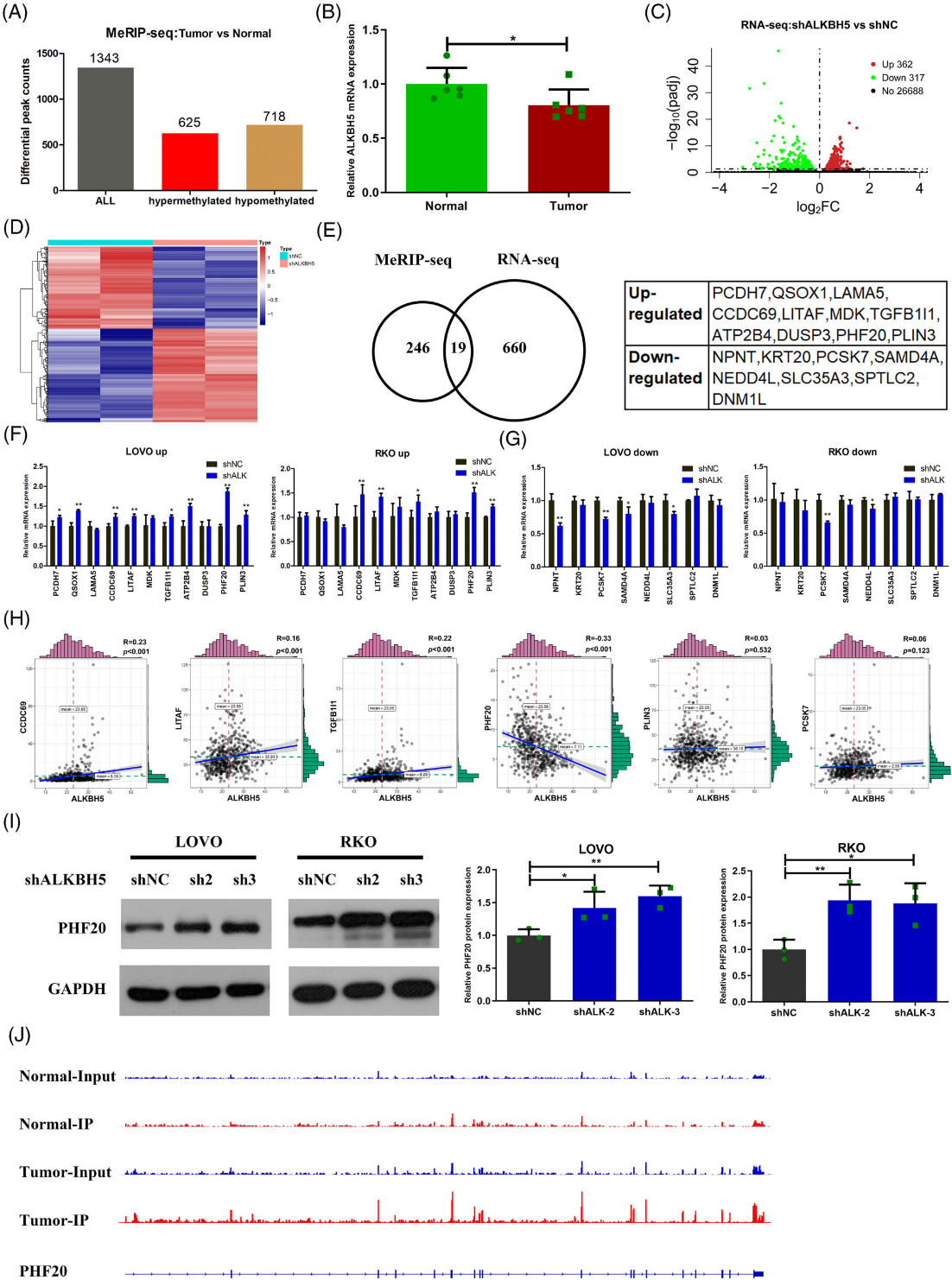
為了鑒定m6A修飾的靶點,我們收集了六名CRC患者的腫瘤組織和正常組織,用于MeRIP-seq。我們通過MeRIP-seEq在六對組織中鑒定了1343個調節失調的m6A峰(圖7A)。其中,625個m6A峰在腫瘤組織中被高甲基化,而718個m6A峰在腫瘤中被低甲基化(圖7A)。接下來,625個高甲基化m6A峰分布在265個基因的轉錄本中,718個低甲基化m6A峰分布在311個基因轉錄本中。有趣的是,我們發現,在MeRIP序列的六對組織中,腫瘤組織中ALKBH5的表達明顯低于正常組織中的表達(圖7B)。我們假設這265個基因的轉錄由ALKBH5調控。為了進一步探索由ALKBH5調控的m6A修飾靶點,我們在ALKBH55敲除的LOVO細胞和對照細胞中進行了RNA序列分析。結果表明,ALKBH5敲除后,679個基因差異表達,包括362個上調基因和317個下調基因(圖7C,D)。結合MeRIP-seq和RNA-seq,我們發現19個基因通過m6A修飾潛在地受ALKBH5調控,包括11個上調基因(PCDH7、QSOX1、LAM5、CCDC69、LITAF,MDK, TGFB1I1, ATP2B4, DUSP3, PHF20和PLIN3) 和8個下調基因(NPNT、KRT20、PCSK7、SAMD4A、NEDD4L、SLC35A3、SPTLC2和DNM1L)(圖7E)。然后,我們檢測了這19個基因在ALKBH5敲除后的表達情況。RT-qPCR結果顯示,11個上調基因中,5個基因(CCDC69、LITAF、TGFB1I1、PHF20和PLIN3)在ALKBH5基因敲除的LOVO和RKO細胞中均上調(圖7F)。此外,我們發現,在8個下調基因中,只有PCSK7在ALKBH5敲除的LOVO和RKO細胞中下調(圖7G)。接下來,我們檢查了ALKBH5與TCGA數據庫中六個候選基因之間的相關性。我們的結果表明,在CRC中,只有PHF20與ALKBH5呈負相關(圖7H)。我們還通過Western blot檢測驗證了與對照細胞相比,ALKBH5敲除細胞中PHF20蛋白表達的升高(圖7I)。此外,可視化分析表明,與正常組織相比,PHF20 mRNA的m6A峰在腫瘤組織中更為豐富(圖7J)。因此,PHF20可能是通過m6A修飾由ALKBH5調節的關鍵分子。
6)ALKBH5的缺失增加了PHF20 mRNA的穩定性,進而促進結腸癌的進展
作為去甲基酶,ALKBH5能夠從mRNA中去除m6A修飾。mRNA的m6A定量分析表明,ALKBH5缺失顯著增加了mRNA的m6A修飾水平(圖8A)。MeRIP qPCR分析表明,ALKBH5缺失顯著增加了PHF20 mRNA的m6A修飾水平(圖8B)。接下來,我們利用SRAMP數據庫和和BEMP數據庫預測PHF20 mRNA的m6A位點。我們發現PHF20 mRNA上的m6A甲基化位點主要位于3′UTR,并且有8個可能的m6A甲基化位點。根據PHF20 mRNA上的m6A甲基化位點,我們構建了熒光素酶報告載體(圖8C)。熒光素酶報告分析表明,ALKBH5缺失顯著增強野生型PHF20 3’UTR質粒的表達,但對具有m6A突變位點的PHF20 3’UTR質粒沒有顯著影響(圖8D)。為了評估ALKBH5對PHF20 mRNA穩定性的影響,我們用放線菌素D處理結腸癌細胞,發現ALKBH55缺失改善了PHF20的穩定性并延長了其半衰期(圖8E)。然后我們進行拯救實驗,以確定ALKBH5是否通過調控PHF20影響結腸癌細胞的生物學功能。結果顯示,PHF20敲除抑制了shALKBH5介導的LOVO和RKO增殖、菌落形成、遷移和入侵的增強(圖8F-I)。

結論:
我們確定ALKBH5表達在CRC中下調,并起到關鍵的腫瘤抑制作用。機制上,ALKBH5通過去除m6A修飾抑制PHF20 mRNA的穩定性。我們的研究為ALKBH5介導的m6A修飾的抗癌作用提供了新的見解,并表明靶向ALKBH5介導的PHF20 mRNA的m6A修飾可能是一種有希望的結腸癌干預和治療策略。
參考文獻:
Zhang Z, Wang L, Zhao L, Wang Q, Yang C, Zhang M, Wang B, Jiang K, Ye Y, Wang S, Shen Z. N6-methyladenosine demethylase ALKBH5 suppresses colorectal cancer progression potentially by decreasing PHF20 mRNA methylation. Clin Transl Med. 2022 Aug;12(8):e940. doi: 10.1002/ctm2.940.