






『基金熱點精讀』“炎癥記憶”驅動過敏性哮喘——“代謝-表觀遺傳回路”介導巨噬細胞重編程
傳染性病原體可以“重編程”或“訓練”巨噬細胞及其祖細胞,使其更容易對隨后的損傷作出反應。然而,這種炎癥記憶是否存在于過敏性哮喘等T2炎癥條件中尚未清楚。作者采用HDM(室內塵螨)過敏患者臨床樣本、HDM誘導的小鼠AAI(氣道過敏性炎癥)和體外訓練相結合的方法,探討巨噬細胞在過敏性哮喘中的免疫機制。該研究于2022年6月發表在《The Journal of Allergy and Clinical Immunology》,IF:10.228。
技術路線:
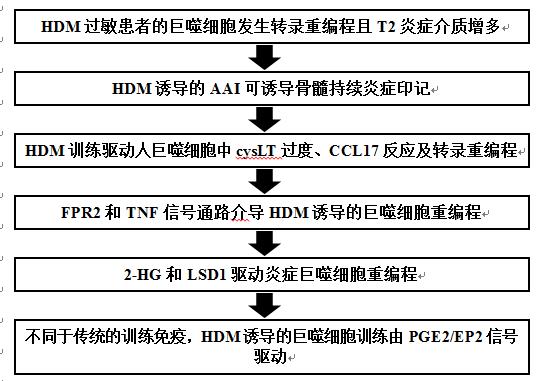
主要研究結果:
1. HDM過敏患者的巨噬細胞發生轉錄重編程且T2炎癥介質增多
為研究過敏性哮喘中潛在的巨噬細胞記憶,作者從HDM過敏或健康供者的單核細胞中產生aMDM(巨噬細胞)。RNA測序分析發現,與非過敏供者相比,HDM過敏供者的aMDM有88個基因差異表達(圖1A和B),表明穩定的轉錄重編程持續整個體外分化。體外HDM暴露導致患者源性aMDM中TNF、IL-12、CXCL2和S100P的產生增加(圖1C),這表明aMDM患者的HDM反應增強以TNF為主。來自HDM過敏個體的未受刺激的aMDM,產生了大量的cysLT,這是T2炎癥的重要介質。哮喘中TH2反應的驅動因子CCL17在HDM患者的aMDM和AM(氣道巨噬細胞)中有升高的趨勢(圖1D和E)。因此,過敏性哮喘患者的aMDM在基線時表現出炎癥印記和T2驅動介質譜,并增強了TNF主導的HDM反應。

圖1 HDM過敏性哮喘患者單核細胞來源的巨噬細胞表現出持續的炎癥基因表達和炎癥介質的過度產生
2. HDM誘導的AAI可誘導骨髓持續炎癥印記
與PBS致敏小鼠相比,BMDM(骨髓起源的巨噬細胞)和AM從HDM致敏小鼠的骨髓祖細胞分化7天,顯示cysLTs產生增加和Ccl17表達增強(圖2A和B)。攻擊7天后,HDM誘導的AAI和T2細胞因子在骨髓中的表達基本消失(圖2C)。然而,AM和BMDM保持了CCL17的高產量(圖2D)。此外,BMDM上調了經典訓練免疫基因(Il6和Ptgs2) (圖2E)。總之,這表明AAI在局部和骨髓祖細胞中都留下了先天記憶。
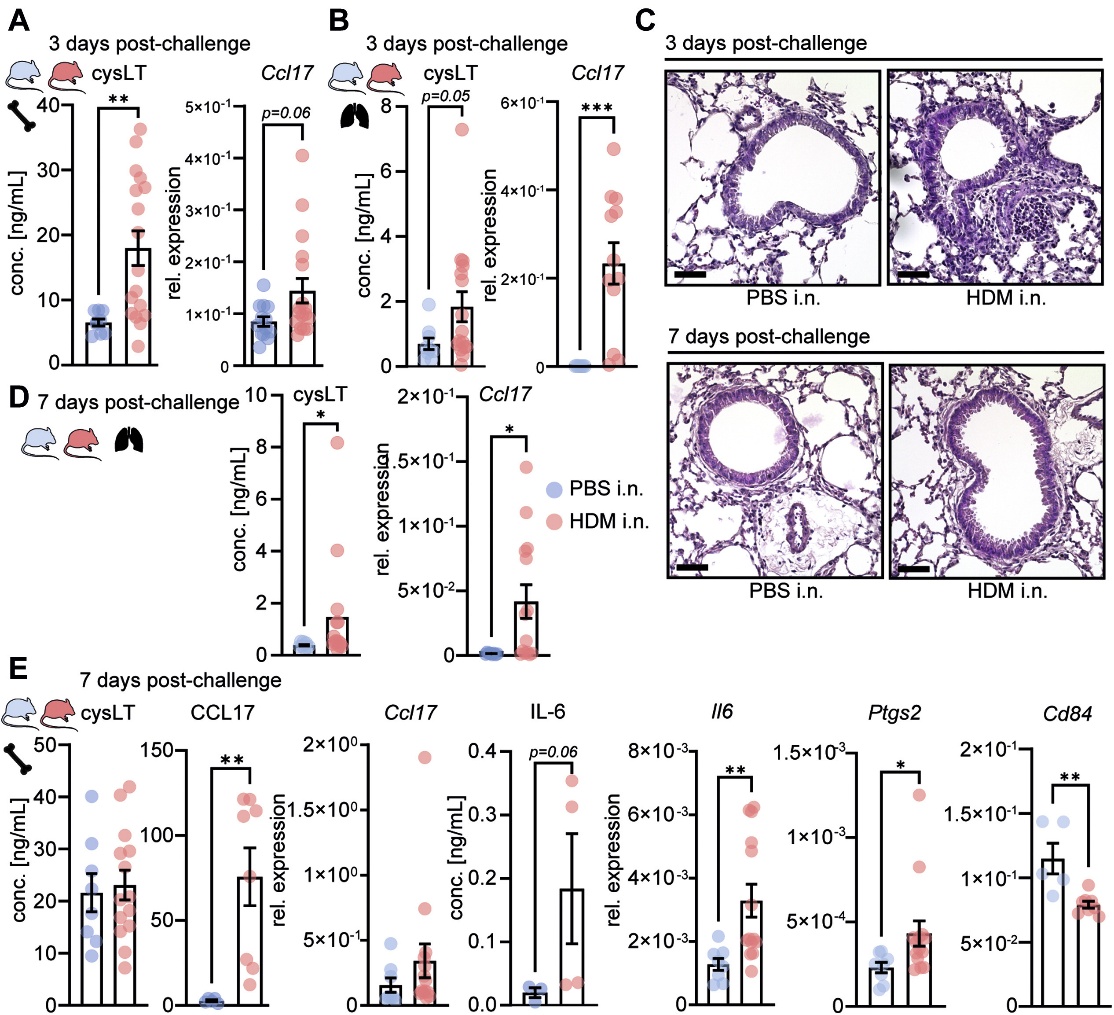
圖2 HDM誘導的氣道炎癥在小鼠外周和AMs中誘導T2印記,向經典的中樞訓練免疫轉移
3. HDM訓練驅動人巨噬細胞中cysLT過度、CCL17反應及轉錄重編程
在分化第7天,用HDM訓練aMDM,經過5天的沖洗期后重新刺激,體外HDM訓練和再刺激的aMDM中cysLT和CCL17的產生增加(圖3A和B)。通過RNA測序,用火山圖和聚類熱圖顯示炎癥基因的差異表達(圖3C、D、E和F)。炎癥基因的表達伴隨HDM訓練巨噬細胞的代謝激活(圖3G、H和I),表明清除HDM后代謝重編程仍然存在。IL17RB(受體亞基結合IL-25)在體外訓練和患者的aMDM中上調(圖1A和B;圖3J)。與對照組aMDM相比,暴露于IL-25的過敏原訓練中,CCL17和cysLT的產生增加(圖3K和L),表明對上皮信號的反應性增強。相反,來自HDM訓練和攻擊的巨噬細胞的上層清液上調人支氣管上皮細胞中的CXCL8(圖3M)。因此,體外HDM訓練誘導哮喘患者巨噬細胞的轉錄和代謝重編程,再現炎癥記憶特征,并對氣道上皮細胞產生功能性影響。

圖3 HDM訓練分化的人類巨噬細胞驅動T2促進和代謝激活表型
4. FPR2和TNF信號通路介導HDM誘導的巨噬細胞重編程
接下來,作者試圖確定HDM介導巨噬細胞重編程的機制。與HDM傳感有關的FPR2,在HDM訓練的巨噬細胞中持續上調,并由HDM刺激誘導(圖4A)。在HDM訓練期間,通過藥物抑制劑(PBP10)阻斷FPR2信號通路可以抑制CCL17的增強反應(圖4B),并阻止TNF的誘導(圖4C),表明FPR2可能是主要的HDM受體,參與HDM驅動的巨噬細胞重編程。作者在HDM訓練期間中和了TNF,從而抑制了HDM刺激的aMDM中CCL17的增強反應(圖4E)。為了測試體內TNF信號傳導的相關性,作者在致敏和攻擊過程中向HDM致敏小鼠注射依那西普(圖4F,頂部)。依那西普治療減弱了來自HDM致敏小鼠的BMDM中CCL17的增加釋放(圖4H,左)。在體外HDM誘導的AAI期間,依那西普治療可防止HDM再刺激來自HDM致敏小鼠的BMDM中CCL17和IL-6的增強反應(圖4H,右;圖4I)。TNF(LysM-cre Tnffl/fl)骨髓缺乏小鼠的致敏和挑戰(圖4F,底部)導致氣道嗜酸性粒細胞減少(圖4J)以及BMDM在基線和IL-4刺激后產生的CCL17減少(圖4K),支持骨髓來源的TNF的作用在HDM誘導的AAI期間骨髓中的T2印記。總之,這表明在過敏原驅動的炎癥過程中,通過FPR2誘導的自分泌TNF信號通路驅動促炎巨噬細胞的記憶。
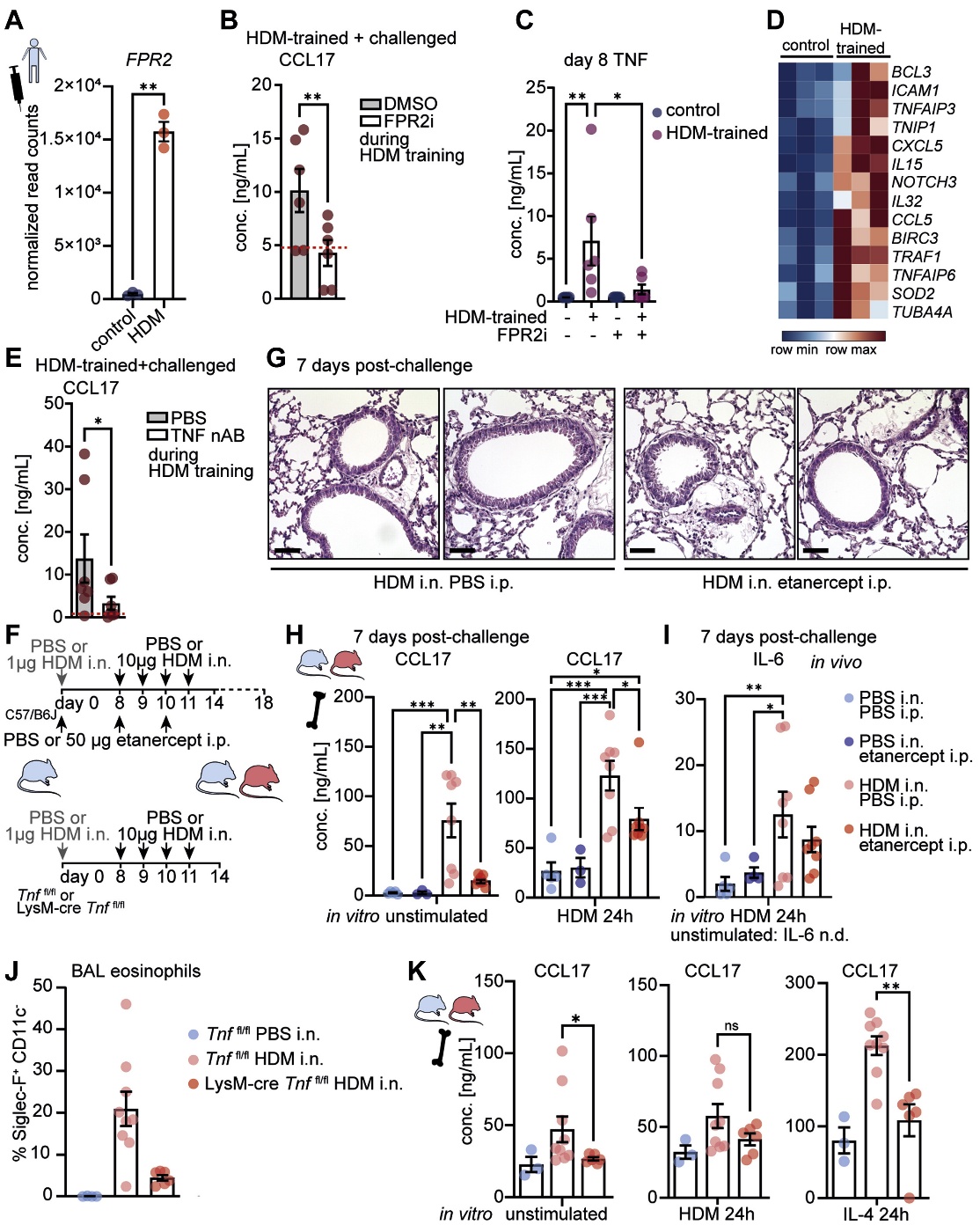
圖4 在體外和體內,自分泌TNF信號通路介導HDM驅動T2印跡
HDM致敏小鼠的BMDM顯示氨基酸和三羧酸循環中間體的輸出增加,包括參與LT生物合成、M2活化和T2免疫的代謝產物(圖5A、B和C)。2-HG是a-酮戊二酸依賴型雙加氧酶活性的調節劑,其產生增加(圖5D)。在BMDM中Arg1表達增加(圖5E、F),表明TNF對T2炎癥的負調節因子具有抑制作用。與HDM致敏的BMDM中2-HG增加一致,急性HDM暴露上調人aMDM中的2-HG(圖5G)。在訓練期間用2-HG替代HDM導致CCL17增強(圖5H),但cysLT對HDM挑戰的反應沒有增強,部分模仿了HDM誘導的訓練。在對巨噬細胞的急性激活過程中添加LPS時,2-HG可增強CCL17、IL1B和PTGS2的誘導(圖5I),表明2-HG可增強aMDM的炎癥激活。在BMDM中,添加2-HG會增加PGE2和CCL17的產生(圖5J),表明2-HG參與T2印記。HIF-1α靶基因,以及HIF1A轉錄在人類巨噬細胞中由HDM誘導(圖5K),但在HDM訓練期間抑制HIF-1α只能部分消除CCL17的增強反應(圖5L)。在訓練過程中KDM1A抑制劑pargyline的應用抑制了CCL17和cysLT對HDM攻擊的反應(圖5M),這表明LSD1介導的重編程是HDM訓練的表觀遺傳機制。
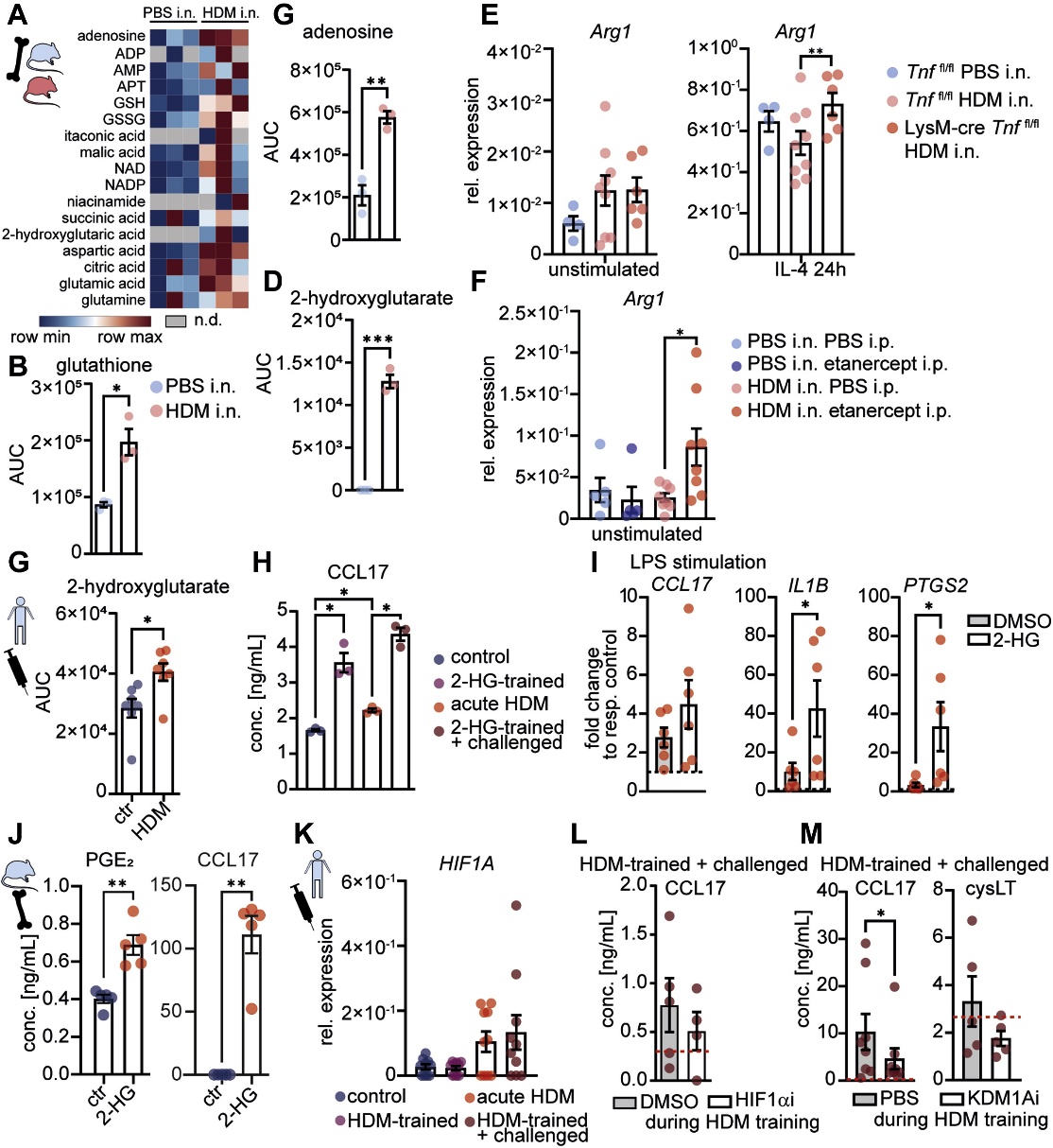
圖5 2-HG和KDM1A介導的“代謝-表觀遺傳”crosstalk促進HDM誘導的巨噬細胞高反應性
6. 不同于傳統的訓練免疫,HDM誘導的巨噬細胞訓練由PGE2/EP2信號驅動
為進一步識別TNF驅動的巨噬細胞代謝和表觀遺傳重編程的下游介質,作者在過敏原訓練后(第8天)、休息5天后(第13天),以及HDM挑戰后24小時(第14天),對HDM訓練的aMDM進行了RNA測序(圖6A和B)。HDM訓練的aMDM在攻擊時也合成了大量的前列腺素(圖6D),并且參與PGE2生成的酶,特別是微粒體前列腺素E合酶1,持續誘導HDM訓練和攻擊(圖6E和F)。與HDM誘導的環氧合酶2一起,這可能解釋了HDM經歷的人和鼠巨噬細胞中HDM觸發的PGE2產生增加的原因(圖6G和H)。依那西普治療后HDM觸發的COX-2(Ptgs2)誘導減少(圖6I),進一步暗示了TNF驅動的重編程中的COX-2/PGE2途徑。與野生型BMDM相比,EP2缺陷型BMDM表現出完整的HDM觸發的TNF反應,但CCL17反應降低(圖6J和K)。表明PGE2增強巨噬細胞的合成代表了TNF介導的先天免疫訓練的下游機制。因此,HDM訓練的巨噬細胞的花生四烯酸代謝增加有助于TNF介導的訓練T2免疫。總之,這些數據確定了導致過敏性哮喘中持續T2炎癥巨噬細胞重編程的代謝-表觀遺傳回路。
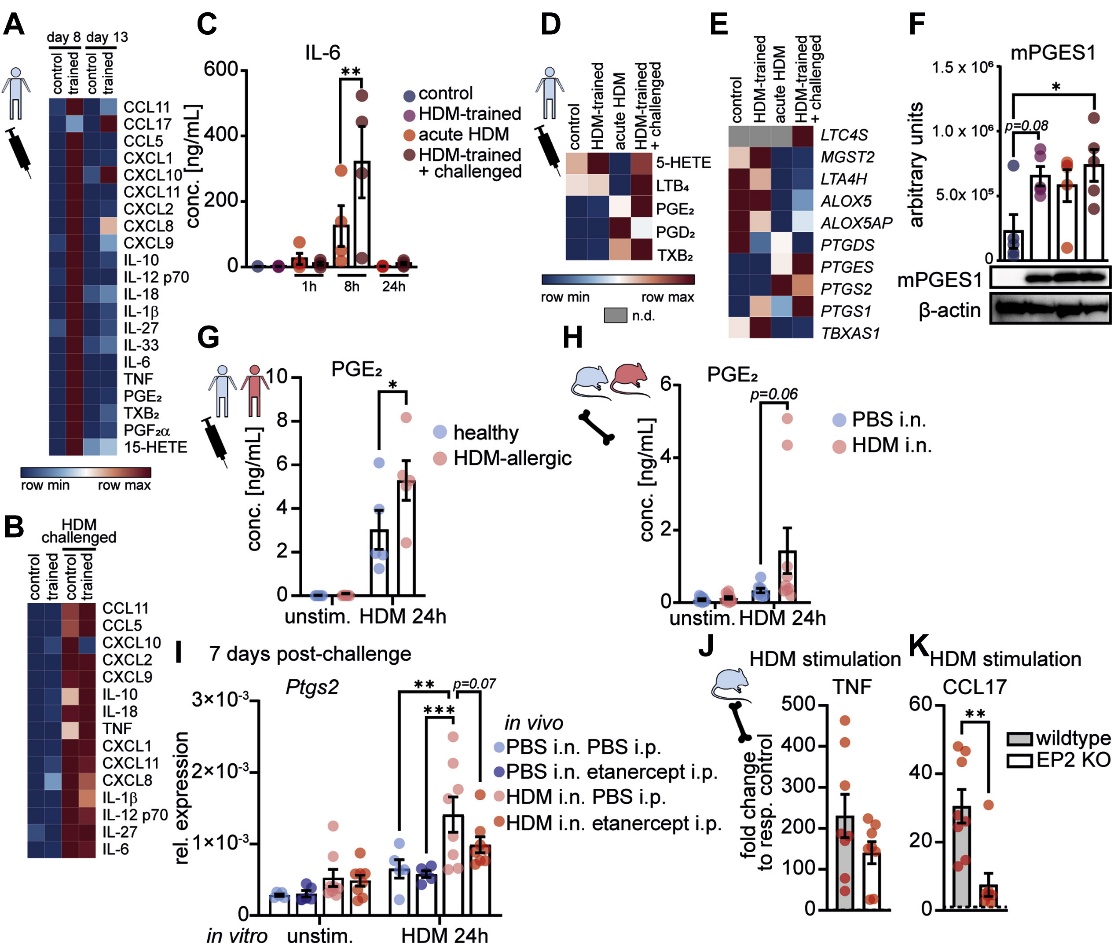
圖6 不同于傳統的訓練免疫,HDM誘導的巨噬細胞訓練由PGE2/EP2信號驅動
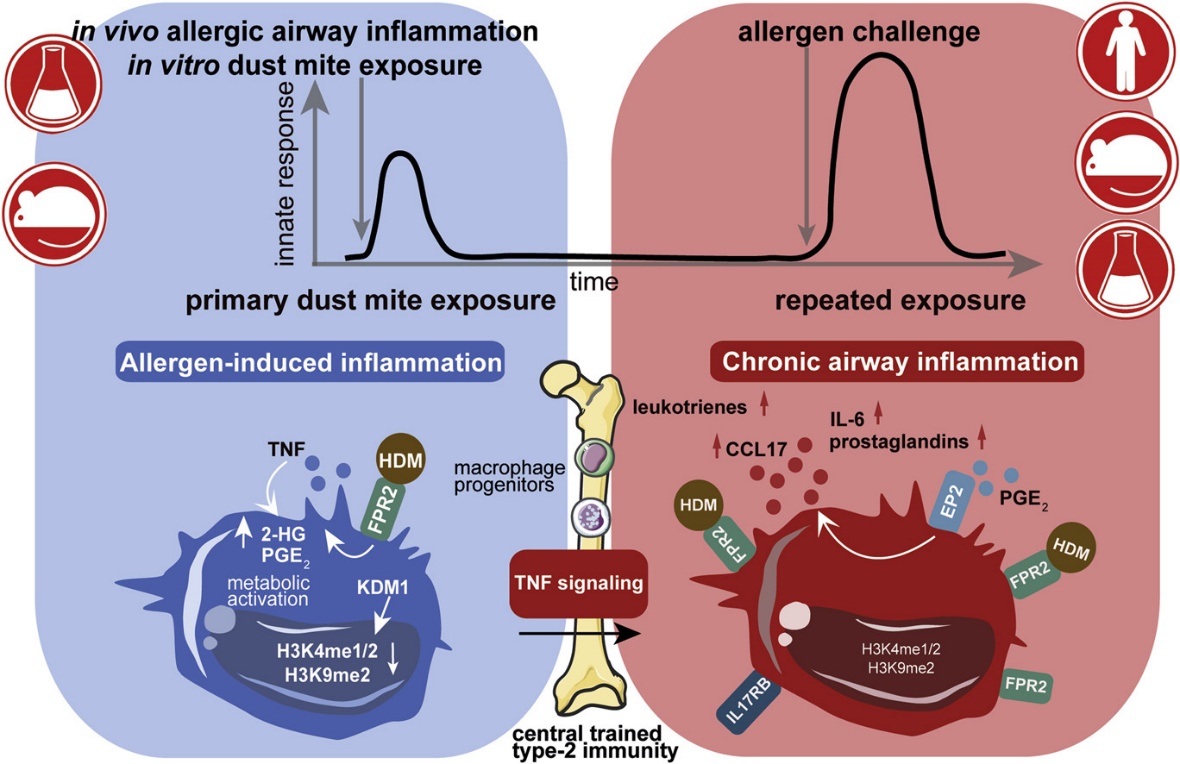
摘要圖
結論:
過敏原引發的炎癥驅動依賴于TNF的先天記憶,這可能會使慢性T2氣道炎癥持續存在并加劇,因此成為哮喘治療的靶點。