






lncRNA NEAT1通過調節miR-362-3p/MIOX軸促進肝細胞癌鐵死亡
lncRNA NEAT1參與調節細胞周期、增殖、凋亡和腫瘤細胞的遷移。然而,NEAT1在調節腫瘤中鐵死亡中的功能和分子機制尚不清楚。在這里,我們發現鐵死亡誘導劑erastin和RSL3通過促進p53與NEAT1啟動子的結合來增加NEAT1的表達。誘導NEAT1通過競爭性結合miR-362-3p促進MIOX的表達。MIOX增加了ROS的產生,降低了細胞內NADPH和GSH的水平,導致erastin和RSL3誘導的鐵死亡增強。重要的是,NEAT1的過表達通過增強體外和體內的鐵死亡,提高了erastin和RSL3的抗腫瘤活性。總的來說,NEAT1通過調節miR-362-3p和MIOX在鐵死亡中發揮了新的、不可或缺的作用。考慮到HCC患者對化療和免疫治療不敏感的臨床發現,對于NEAT1高表達的HCC患者,誘導鐵死亡可能是一種有希望的治療策略。本文于2022年3月發表于Cell Death and Differentiation(IF=15.828)上。
技術路線

結果
1)鐵死亡誘導NEAT1表達依賴于p53
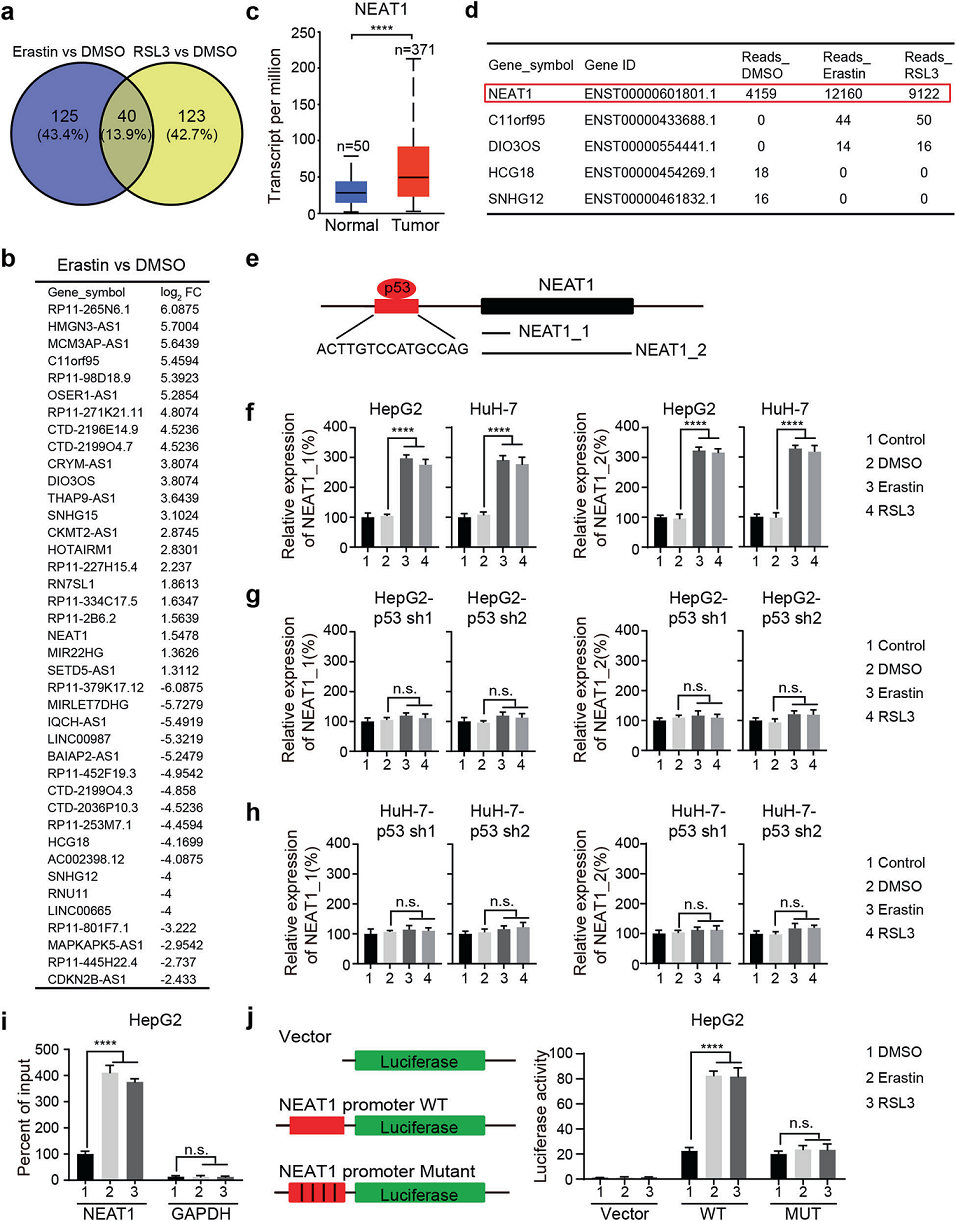
為了鑒定與鐵死亡有關的lncRNAs,用鐵死亡誘導劑處理肝癌細胞系HepG2,用RNA-Seq進行轉錄組分析。如圖所示,在erastin和RSL3處理后,40個lncRNAs導致兩種處理的基因表達發生顯著變化,以及分別受erastin或RSL3影響的125或123個lncRNAs(圖1a、b)。我們根據差異表達基因列表挖掘了UALCAN數據庫,發現了五種有趣的lncRNAs(NEAT1、C11orf95、DIO3OS、HCG18、SNHG12),它們在HCC中的表達與正常組織相比顯著(圖1c)。其中,NEAT1的表達顯著高于其他lncRNAs(圖1d)。因此,我們進一步研究NEAT1在鐵死亡中的作用和機制。
NEAT1有兩種亞型,NEAT1_1在大多數人體組織中組成性表達,而NEAT1_2的表達是組織特異性的。然而,NEAT1的生物學功能主要來源于NEAT1_2亞型。為了檢測兩種亞型的表達,我們設計了引物來產生兩個擴增子,引物1檢測到NEAT1_1和NEAT1_2的表達,引物2僅檢測到NEAT1_2(圖1e)。如圖1f所示,在erastin和RSL3處理的HCC細胞中,這兩種轉錄物均上調,表明NEAT1_1和NEAT1_2的表達在鐵死亡中被誘導。另外,我們發現,p53的缺失消除了HepG2和HuH-7細胞中NEAT1的上調(圖1g,h),表明erastin和RSL3誘導的NEAT1表達是由p53介導的。為了進一步測試p53是否直接調節NEAT1的轉錄,我們使用JASPAR程序分析了NEAT1的啟動子區域,并發現了一個p53的結合序列。然后,我們進行ChIP分析,發現erastin和RSL3處理促進了p53與NEAT1啟動子區域的結合(圖1i)。此外,我們觀察到在erastin或RSL3處理NEAT1-WT啟動子后,啟動子活性增加(圖1j)。綜上所述,在鐵死亡誘導條件下,p53直接結合NEAT1的啟動子并促進NEAT1的表達。
2)NEAT1促進erastin和RSL3誘導的鐵死亡
為了研究NEAT1是否調節鐵死亡,我們構建了兩個NEAT1特異性shRNA。NEAT1 shRNA 1同時敲除NEAT1_1和NEAT1_2,而NEAT1 shRNA 2僅敲除NEAT1_2,而不影響NEAT1_1的表達(圖2a、b)。接下來,我們建立了穩定表達兩種不同NEAT1特異性shRNA的HepG2和HuH-7細胞系,并用erastin或RSL3處理它們。如圖2c所示,在HepG2或HuH-7細胞中敲除NEAT1亞型或單獨敲除NEAT1_2可顯著抑制erastin和RSL3誘導的細胞死亡。NEAT1敲除顯著抑制MDA、脂質ROS和Fe2+的積累(圖2d、e、f)。總的來說,NEAT1促進erastin和RSL3誘導的肝癌細胞鐵死亡。

3)NEAT1通過調節MIOX的表達來調節鐵死亡
為了進一步闡明NEAT1在鐵死亡中的作用機制,我們通過比較敲除NEAT1導致的基因表達差異來尋找NEAT1靶基因。如圖3a和b所示,在轉染shRNA對照的HepG2細胞中,經erastin處理后,有1953個上調基因和2098個下調基因。然而,在轉染NEAT1 shRNA的HepG2細胞中,599個上調基因和711個下調基因對erastin無反應,表明這些基因在鐵死亡中的修飾表達依賴于NEAT1(圖3a,b)。此外,我們鑒定了6個上調基因和15個下調基因,它們具有更顯著的修飾(圖3c),并發現在erastin處理后,MIOX是上調最高的基因。同時,qRT-PCR檢測表明,erastin和RSL3均不能誘導NEAT1敲除細胞中MIOX的上調,這進一步證實了鐵死亡中MIOX表達的誘導依賴于NEAT1(圖3d)。MIOX過表達拯救了由NEAT1缺乏抑制的erastin和RSL3誘導的鐵死亡(圖3e)。MIOX過表達增加NEAT1敲除細胞內MDA、脂質ROS和Fe2+水平(圖3f-h)。總之,這些結果表明NEAT1通過調節MIOX的表達促進erastin和RSL3誘導的鐵死亡。

4)MIOX通過調節Fe2+、GSH和NADPH促進鐵死亡
先前的一項研究表明,MIOX過表達可通過調節鐵積累、GSH活性和NADPH水平,加重順鉑誘導的急性腎損傷。為了測試這種機制在erastin和RSL3誘導的HCC細胞鐵死亡中是否保守,我們在HCC細胞中過表達MIOX,并發現MIOX促進erastin和RSL3誘導的細胞死亡(圖4a)。相反,敲低MIOX可抑制erastin和RSL3誘導的細胞死亡(圖4e)。我們進一步檢測了細胞內的Fe2+水平。如圖4b所示,經過erastin和RSL3處理后,MIOX過表達的細胞內鐵濃度顯著升高。在用erastin治療后,MIOX的過表達進一步降低了HepG2和HuH-7細胞中的GSH水平(圖4c)。同樣,NADPH有助于消除脂質ROS,并調節細胞對鐵死亡的敏感性,在MIOX過表達的細胞中,NADPH也會降低(圖4d)。相反,在HepG2和HuH-7細胞中,耗盡MIOX抑制了erastin誘導的HepG2和HuH-7細胞中Fe2+的積累、NADPH的降低和GSH的降低(圖4f-h)。總之,MIOX通過調節細胞內的Fe2+、GSH和NADPH水平促進鐵死亡(圖4i)。

5)MiR-362-3p參與NEAT1對MIOX的調節
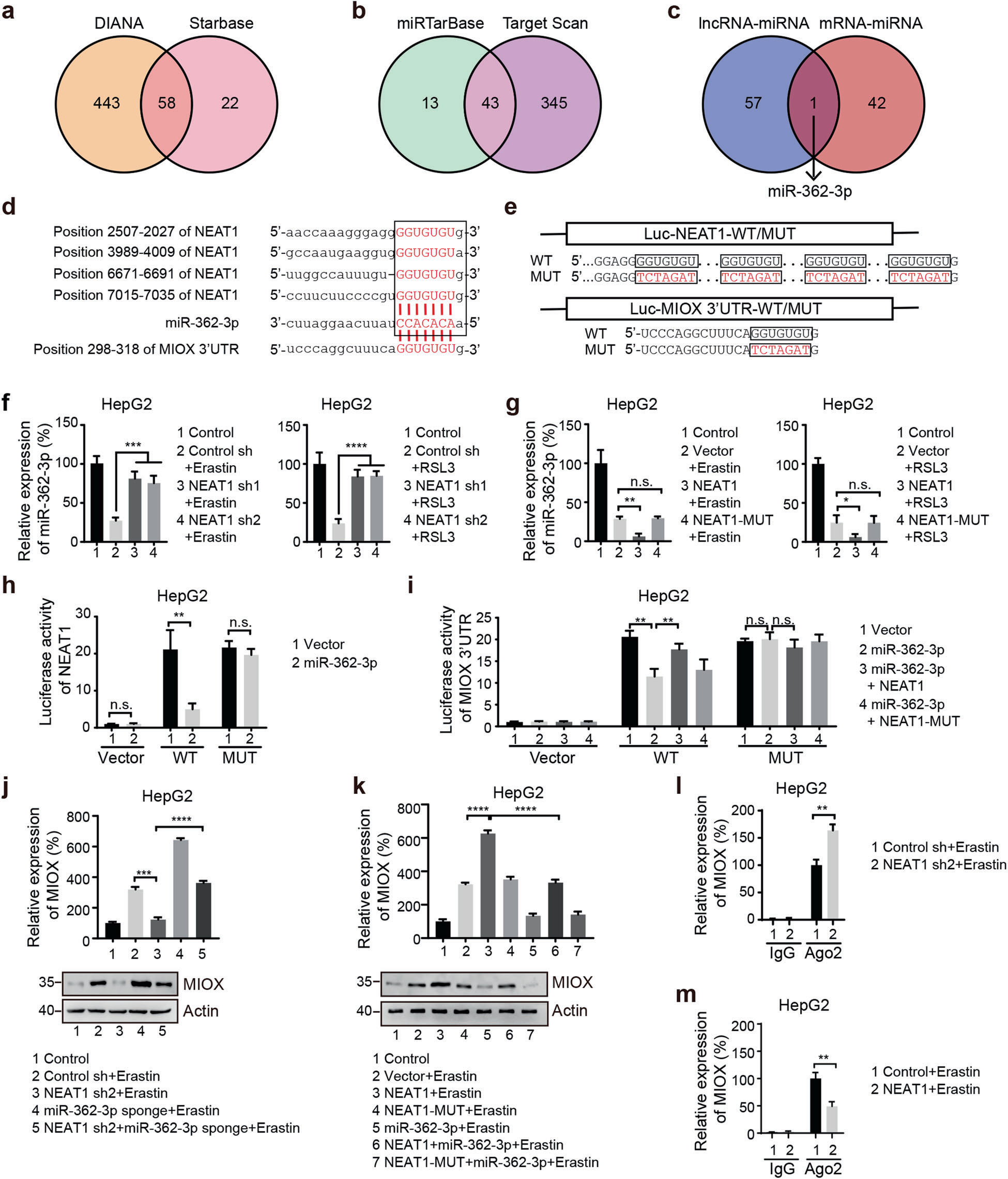
越來越多的證據表明,lncRNAs通過與miRNAs(如ceRNA)結合來調節靶基因。為了確定NEAT1在鐵死亡期間是否作為ceRNA發揮作用,我們使用預測工具,使用starbase v2.0和DIANA-LncBase V2,在NEAT1中找到58個潛在的miRNA結合位點(圖5a)。由于MIOX被證明是鐵死亡中NEAT1的靶點,我們隨后使用miRTarBase和targetscan在MIOX中尋找潛在的miRNA結合位點,兩個數據庫中都存在43個潛在的MIOX結合miRNA(圖5b)。通過比較兩種方法預測的miRNA,我們發現miR-362-3p是唯一重疊的miRNA(圖5c),它可能通過NEAT1介導MIOX和鐵死亡的調節。使用兩種公開的生物信息學工具targetscan和starbase,我們發現NEAT1和MIOX的3′UTR包含假定的miR-362-3p結合位點(圖5d)。接下來,我們進行熒光素酶活性測定(圖5e)。如圖5f 所示,在erastin或RSL3處理后,NEAT1的敲除增加了HepG2和HuH-7細胞中miR-362-3p的表達水平。相反,NEAT1的過表達降低了miR362-3p的表達水平(圖5g)。miR-362-3p的過表達顯著抑制野生型NEAT1的轉錄活性(圖5h)。總之,這些結果表明NEAT1調節miR-362-3p。
為了確定MIOX是否是miR-362-3p的直接靶點,我們將miR-362-3p過表達載體與包含MIOX的3′UTR的報告載體共同轉染。如圖5i所示,miR-362-3p的過表達顯著減弱了含有MIOX野生型3′UTR的報告載體的熒光素酶活性。相反,序列的突變阻斷了miR-362-3p的抑制作用(圖5i)。此外,我們發現miR-362-3p對MIOX 3’UTR活性的影響可以通過過表達的NEAT1部分恢復(圖5i)。為了進一步證明miR-362-3p參與MIOX和NEAT1之間的交叉調節,我們評估了MIOX的表達。在erastin或RSL3處理的HepG2細胞中,NEAT1敲除抑制了MIOX的mRNA和蛋白質表達。相反,miR-362-3p的敲除顯著增加了被NEAT1 shRNA抑制的MIOX的mRNA和蛋白質表達(圖5j)。此外,野生型NEAT1的過表達增加了MIOX的mRNA和蛋白質表達。miR-362-3p的過表達顯著抑制NEAT1誘導的MIOX的mRNA和蛋白質表達(圖5k)。miRNA以Ago2依賴的方式抑制mRNA的翻譯和降解。為了測試MIOX和miR-362-3p之間的相互作用是否受到NEAT1的影響,對NEAT1過表達或敲除的HepG2細胞進行RIP實驗。如圖5l所示,被Ago2-miR-362-3p復合物拉下的MIOX mRNA水平在NEAT1缺失的HepG2細胞中顯著富集,而過表達NEAT1則顯著降低MIOX mRNA水平(圖5m)。總之,這些結果表明NEAT1通過miR-362-3p積極調節MIOX表達。
6)MiR-362-3p通過調節MIOX抑制鐵死亡
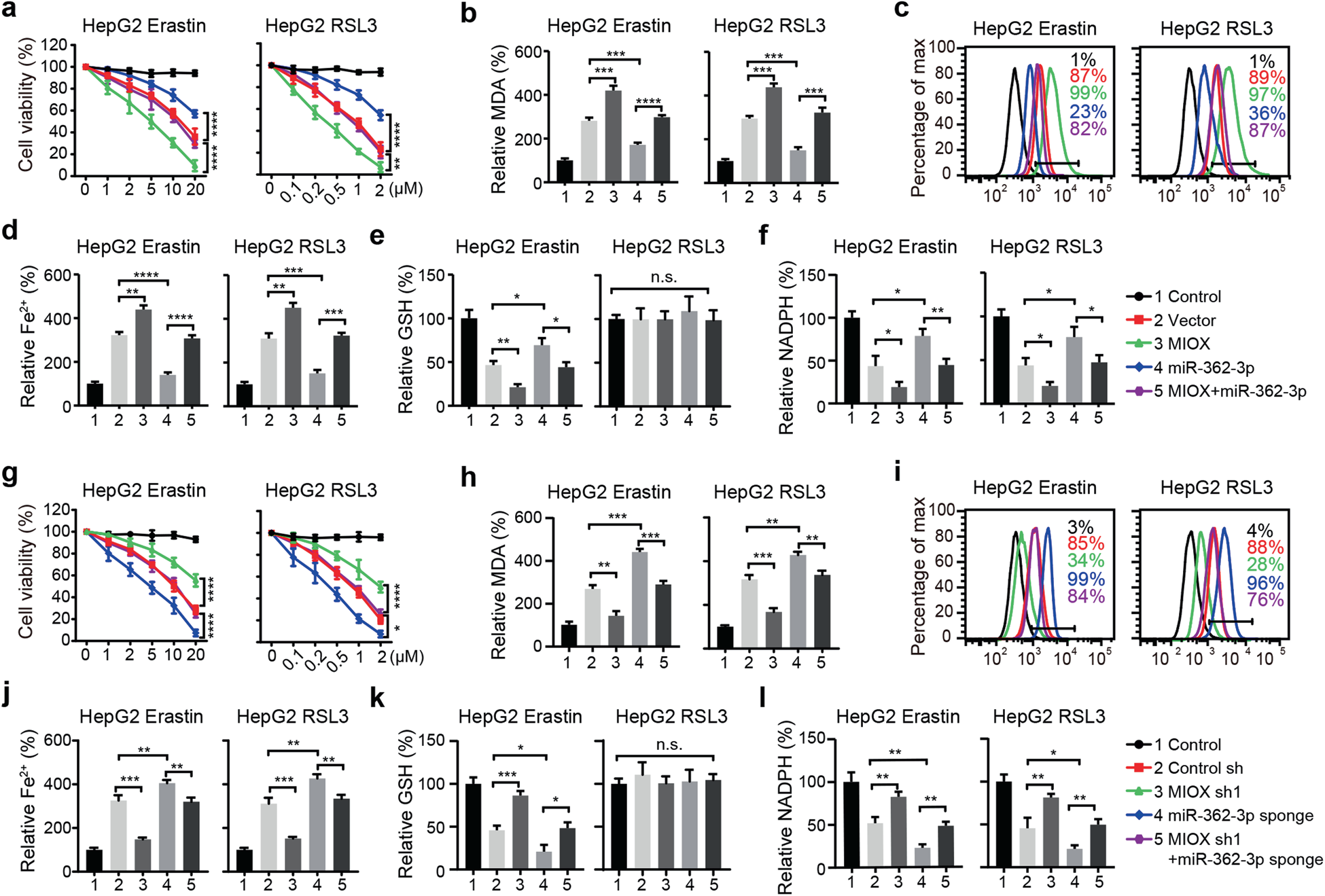
為了分析miR-362-3p是否通過對MIOX的影響來調節鐵死亡,我們通過過表達MIOX進行了拯救實驗。轉染miR-362-3p的HepG2細胞顯著抑制erastin和RSL3誘導的細胞死亡,并降低MIOX的促進作用(圖6a)。同時,過表達MIOX后,MDA、脂質ROS和Fe2+的積累也部分恢復(圖6b-d)。此外,MIOX的過表達抑制了由miR-362-3p誘導的GSH和NADPH的細胞內水平(圖6e,f)。轉染miR-362-3p海綿的HepG2細胞顯著促進了erastin-和RSL3-誘導的細胞死亡和相關的鐵死亡,包括脂質ROS產生、鐵積累、NADPH抑制和erastin誘導的GSH缺失(圖6g-l)。MIOX的下調抑制了由miR-362-3p海綿促進的erastin-和RSL3誘導的鐵死亡(圖6g-l)。這些結果表明miR-362-3p通過調節MIOX的表達來調節鐵死亡。
7)NEAT1過表達促進erastin或RSL3在體內外誘導的鐵死亡
我們通過慢病毒載體在HepG2和HuH-7細胞中穩定地過表達NEAT1,用erastin或RSL3處理細胞,然后通過集落形成測定其對細胞活力的影響。我們發現NEAT1高表達水平的細胞對erastin和RSL3誘導的鐵死亡細胞死亡更敏感(圖7a )。進一步探討NEAT1表達水平對erastin或RSL3體內抗癌活性的影響。將NEAT1穩定過表達的HepG2和HuH-7細胞皮下注射到NU/NU裸鼠體內。NEAT1表達增加的腫瘤的大小明顯小于使用相同藥物治療的對照組(圖7b、c),并且表現出MDA水平升高和erastin誘導的GSH水平降低(圖7d、e)。此外,在NEAT1過表達細胞中用erastin或RSL3處理后,4-HNE的染色顯著增加(圖7f)。同樣地,在NEAT1過表達細胞中,MIOX表達顯著升高。ISH染色證實,erastin或RSL3處理后,NEAT1過表達細胞中miR-362-3p的表達降低(圖7f)。總之, NEAT1調節了erastin和RSL3在HCC細胞中的抗腫瘤活性。
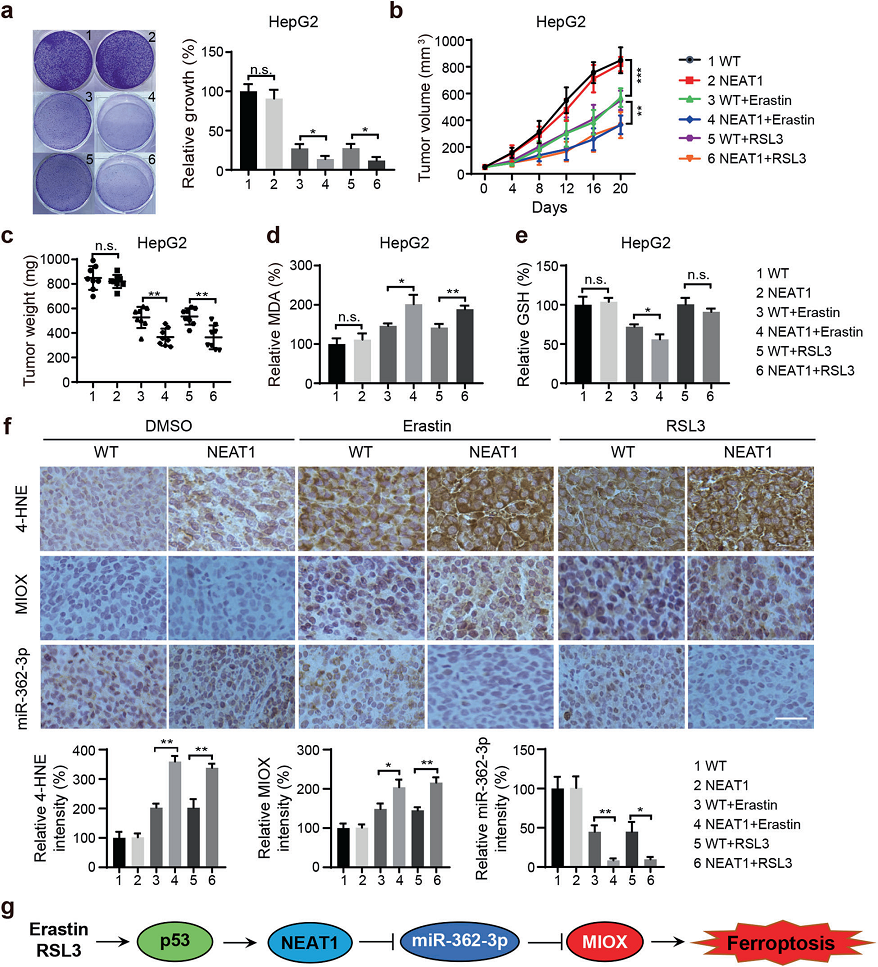
結論:我們的研究結果概述了NEAT1在鐵死亡中的關鍵作用及其調節機制,表明對于高表達NEAT1的腫瘤患者,誘導鐵死亡可能是一種有前途的治療策略。
參考文獻:
Zhang Y, Luo M, Cui X, O'Connell D, Yang Y. Long noncoding RNA NEAT1 promotes ferroptosis by modulating the miR-362-3p/MIOX axis as a ceRNA. Cell Death Differ. 2022 Mar 25. doi: 10.1038/s41418-022-00970-9. Epub ahead of print. PMID: 35338333.