






心律失常性心肌病的潛在新療法:crenolanib藥物抑制PDGFRA
FLNC截斷突變(FLNCtv)是遺傳性擴張型心肌病(DCM)的常見病因,是發展成心律失常性心肌病的高危因素。本研究結果表明,PDGFRA信號通路與DCM發病機制有關,抑制該通路可能是flnc相關心肌病的一種治療策略本研究于2022年2月發表在《Science Advances》IF:14.136雜志上。
技術路線:

主要研究結果:
1、使用患者特異性多能干細胞來源的心肌細胞(iPSC-CMs)構建FLNC-相關DCM模型
作者從FLNCG1891Vfs61X和FLNCCE2189X的DCM患者身上分離出iPSC細胞,然后誘導成iPSC-CMs,并探究其電生理學屬性,結果顯示與健康對照組相比,FLNCtv患者的iPSC-CMs均表現出較弱的收縮性和較慢的放松性(Fig. 1 A and B)。兩種患者特異性iPSC-CMs也表現為自發性心律失常(Fig. 1 C)。與對照組iPSC-CMs相比,心律失常表型也表現為心率的較大變異(Fig. 1 D)。為了進一步驗證心律失常的表型,檢測了健康和iPSC-CMs患者的動作電位(AP)形態。與收縮數據一致,40%的FLNCG1891Vfs61X和36%的FLNCCE2189X 的iPSC-CMs與對照iPSC-CMs相比出現異常AP形態(Fig. 1 E and F)。總之,這些數據表明,FLNC患者iPSC-CMs概述了體外與FLNC相關的DCM相關的心律失常表型。

圖1 FLNCG1891Vfs61X and FLNCE2189X iPSC-CMs 表現出收縮性受損和心律失常
2、FLNCG1891Vfs61X和FLNCCE2189X都是缺失突變體其導致FLNC相關心肌病的單倍機能不全
接下來檢測了患者特異性iPSC-CMs中FLNC的表達水平,通過免疫印跡和qPCR檢測發現,與對照組iPSC-CMs相比,FLNCG1891Vfs61X和FLNCCE2189X突變體表達FLNC蛋白和mRNA水平均顯著降低(Fig. 2 A to C)。盡管FLNC表達水平降低,但通過免疫熒光染色和共定位分析,能夠在兩個FLNC突變的iPSC-CM細胞系中檢測到FLNC及其與α- actitin在Z-discz盤的共定位(Fig. 2D)。
為了探究FLNCG1891Vfs61X和FLNCCE2189X是否是功能丟失的突變體,為FLNC缺失的iPSC系和等基因對照系細胞生成了差異表達基因(DEG)譜,并與iPSC- CMs患者的DEG譜進行了比較。結果顯示成功構建了FLNC純合子(FLNCKO?/?)和雜合子(FLNCKO+/?)敲除的iPSC-CMs(Fig. 2 E to H)。FLNCKO?/?和FLNCKO+/?iPSC-CMs中FLNC的表達在mRNA和蛋白水平上均以等位基因依賴的方式顯著降低。RNA測序生物信息學分析顯示,FLNC截斷突變中的DEGs與FLNC KO iPSC-CMs中的DEGs呈正相關,支持功能缺失是FLNC突變引起表型表現的主要機制(Fig. 2I)。此外,攜帶FLNC突變的iPSC-CMs顯著降低了ID蛋白水平;FLNC KO iPSC-CMs也有相同的發現(Fig. 2J)。最后,FLNCKO?/?細胞表現出與患者iPSC-CMs相似的心律失常表型(Fig. 1 E and F)。總之,這些結果說明FLNCG1891Vfs61X和FLNCCE2189X都是缺失突變體其導致FLNC相關心肌病的單倍機能不全。
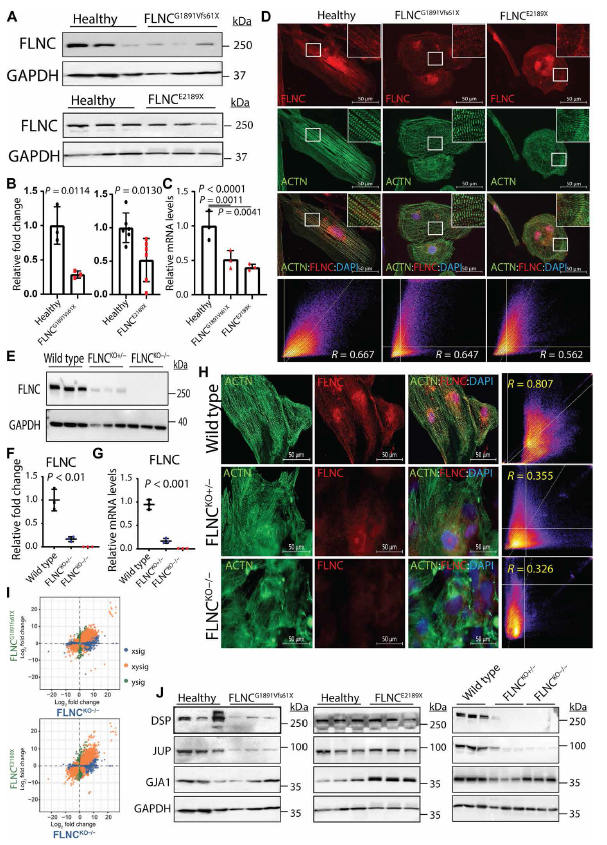
圖2 FLNCG1891Vfs61X and FLNCE2189X的功能缺失突變導致FLNC相關心肌病的單倍機能不全
3、FLNC缺失導致β-Catenin核定位和β-Catenin信號通路的激活
為了發現FLNC的分子伴侶,進行了共免疫沉淀和蛋白質組學分析,通過蛋白質組學分析確定了β-Catenin,經免疫印跡證實(Fig. 3A)。在FLNCKO?/? iPSC-CMs的裂解液中沒有檢測到β-Catenin,而且在FLNC突變的iPSC-CMs的裂解液中β-Catenin的水平也減少了(Fig. 3A)。此外,在FLNC -突變體和FLNCKO?/? iPSC-CMs的細胞核中檢測到大量的β-Catenin,而對照iPSC-CMs中β-Catenin主要在細胞質中檢測到(Fig. 3B-C)。此外,FLNC -突變體和FLNCKO?/? iPSC-CMs的RNA-seq數據表達譜顯示β-Catenin信號通路激活(Fig. 3D)。總之,數據表明β-Catenin可能是一種新的FLNC結合伙伴和FLNC的下游效應物,并且iPSC-CMs中FLNC的缺失導致β-Catenin從胞漿遷移到細胞核,導致β-Catenin信號的激活。
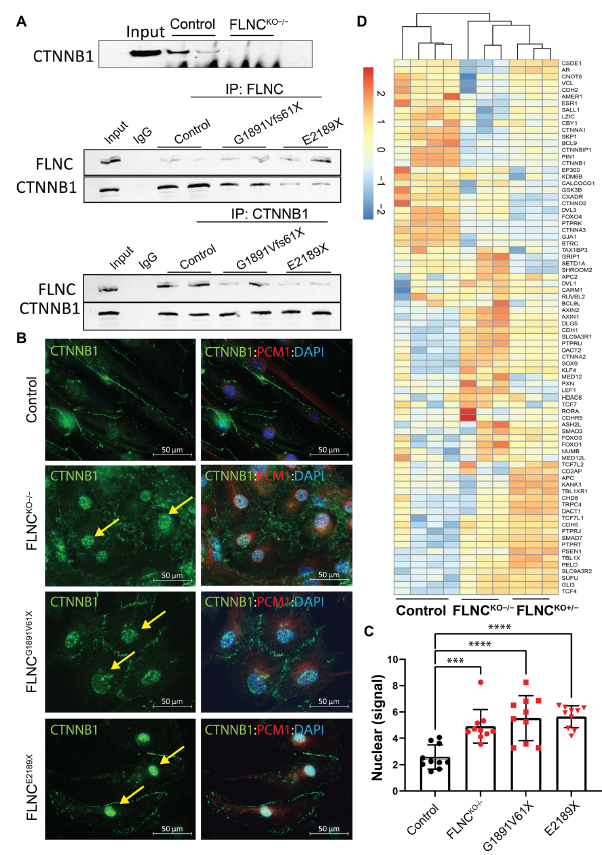
圖3 β-Catenin與FLNC和FLNC激活的β-Catenin信號通路的缺失有關
4、PDGFRA在FLNC相關心肌病中上調并且是β-Catenin的下游調控因子
為鑒定更多的下游靶點用于藥物開發,作者比較了FLNCKO?/?和FLNCKO+/?的iPSC-CMs的轉錄組以鑒定潛在靶基因。如圖Fig. 4A所示,FLNCKO?/?和FLNCKO+/?的iPSC-CMs的轉錄組有接近相似的上調和下調DEGs。對兩者間共有的上調和下調DEGs進行KEGG分析,發現它們主要參與調控cell-cell adhesion, intercellular communication,ion transport等通路(Fig. 4B to D)。此外,這些上調的DEGs主要富集至PDGF分子水平的結合活性(Fig. 4E)。數據顯示,與等基因對照的iPSC-CMs相比,FLNC KO系中PDGFRA的表達水平顯著提高了4倍(Fig. 4F)。由于數據表明,FLNCG1681Vfs61X和FLNCE2189X突變導致單倍功能不全,作者假設攜帶這些突變的iPSC-CMs患者的PDGFRA水平也可能升高。如Fig. 4G to H所示,與對照組相比,兩種患者來源的iPSC-CMs中PDGFRA均上調。總之,這些發現提示PDGFRA信號通路的激活與FLNC相關心肌病的發病機制有關。
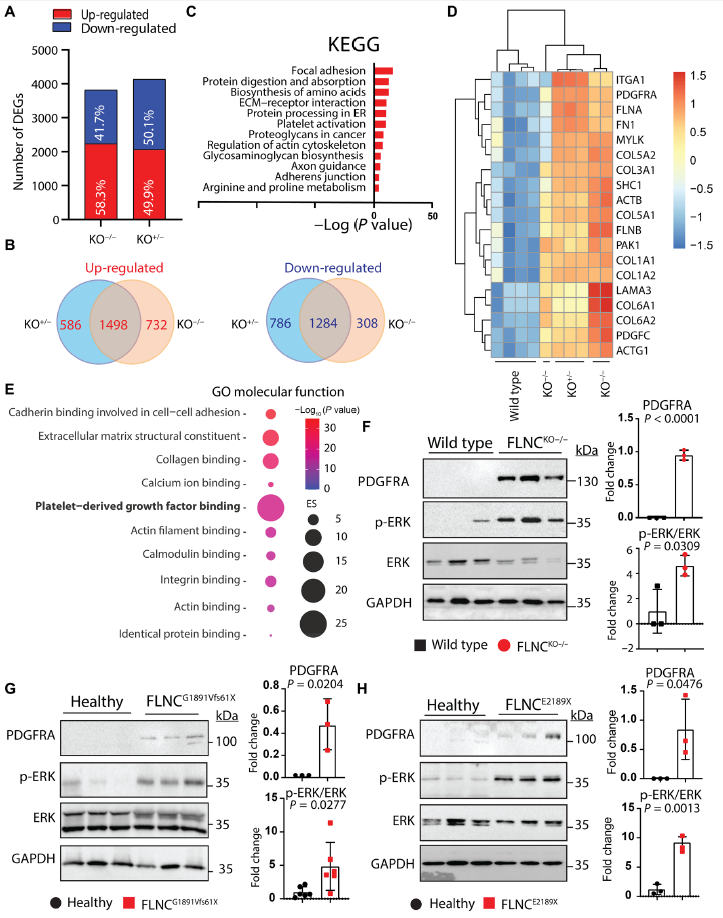
圖4細胞粘附途徑的失調導致PDGFRA在FLNC相關心肌病發病機制中的激活
β-Catenin被認為可以上調PDGFRA在其他組織和/或疾病中的表達。由于觀察到β-Catenin在FLNC缺失的iPSC-CMs中有顯著的核定位,于是研究了β-Catenin是PDGFRA上游轉錄激活因子的假設。因此,使用β-Catenin抑制劑(JW67和JW74)在不同濃度下抑制β-Catenin,并觀察到PDGFRA轉錄水平的劑量依賴性抑制(Fig. 5A)。此外,ChIP下拉實驗表明,已知的轉錄因子和β-Catenin結合伙伴SOX2與PDGFRA啟動子區結合(Fig. 5B)。總之,數據表明β-Catenin是PDGFRA的轉錄激活因子,并且β-Catenin的核定位導致PDGFRA在FLNC缺失的iPSC-CMs中持續上調。
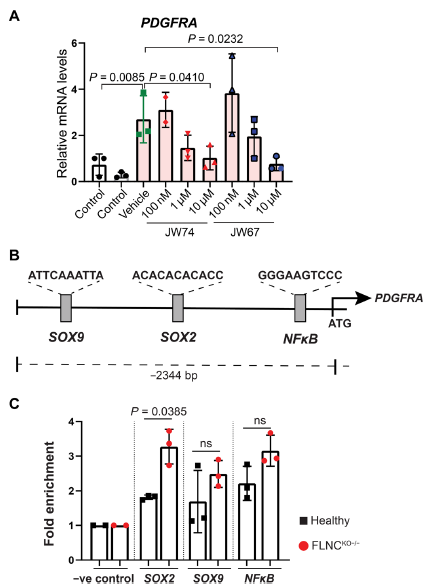
研究顯示PDGFRA通過激活MAPK/ERK信號通路,破壞細胞膜上connexin43(縫隙連接蛋白-1 (GJA1))的信號。作者發現,與對照iPSC-CMs相比,FLNC突變體和KO iPSC-CMs的ERK激活并且GJA1離開細胞膜(Fig. 6A)。為了測試FLNC單倍不足的iPSC-CMs中PDGFRA的激活是GJA1定位錯誤的原因,使用FDA批準的藥物crenolanib對PDGFRA進行藥物抑制,以確定對GJA1膜定位的影響。免疫熒光研究表明,crenolanib抑制PDGFRA可以挽救GJA1在膜上的定位(Fig. 6A)。
轉錄組分析顯示,與FLNCG1891Vfs61X和KO iPSC-CMs相比,FLNCE2189X突變體iPSC-CMs對crenolanib的反應最大,拯救的DEGs數量最多(Fig. 6B)。GSEA揭示了細胞粘附途徑(hsa04510)的部分正常化,支持了cell-cell adhesion位點的結構穩定促進了GJA1粘附在細胞膜上的假設(Fig. 6C)。然而,通過免疫印跡檢測,發現crenolanib處理后GJA1的表達水平沒有改變,這表明PDGFRA抑制并不影響GJA1的表達水平,而是影響GJA1的轉運(Fig. 6D)。最后,也是最重要的一點是,crenolanib處理也顯著改善了來自FLNC患者的iPSC-CMs的收縮性,并減少了心律失常,但對健康對照組而言并非如此(Fig. 6E-F)。綜上,說明FLNC缺失導致細胞-細胞黏附系統失調,抑制PDGFR信號通路改善FLNC突變體iPSC-CMs的收縮性.
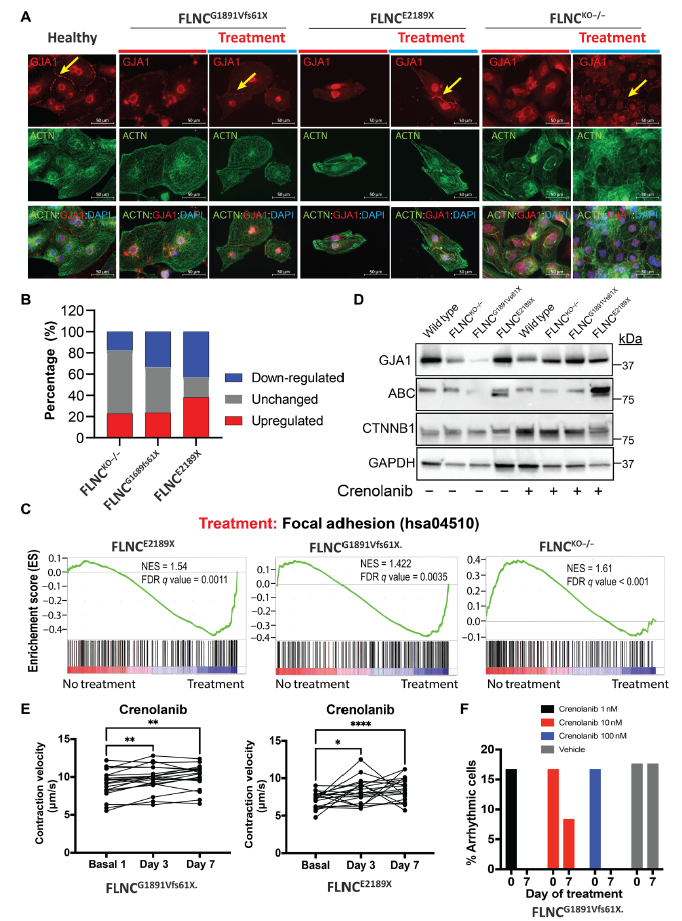
圖6 FLNC缺失導致細胞-細胞黏附系統失調,抑制PDGFR信號通路改善FLNC突變體iPSC-CMs的收縮性
通過免疫熒光檢測到,與對照組iPSC-CMs相比,FLNC突變體iPSC-CMs中β-catenin的核定位增加,但在crenolanib處理后,β-catenin的核定位減少(Fig. 7A)。qPCR證實crenolanib處理對β-catenin核信號轉導的抑制作用,β-catenin靶基因TCF4、SOX9、COL1A2、TGF 3的轉錄水平降低(Fig. 7B)。此外,通過GSEA分析比較crenolanib治療前后患者源性和KO iPSC-CMs的DEGs譜,進一步證實PDGFRA信號抑制后β-catenin信號通路部分恢復(Fig. 7C-D)。因此,證據表明PDGFRA的激活增強了β-catenin信號,并促進了一個正反饋回路。此外,PDGFR抑制可以部分恢復β-catenin膜定位,減少FLNCtv突變引起的β-catenin的激活。
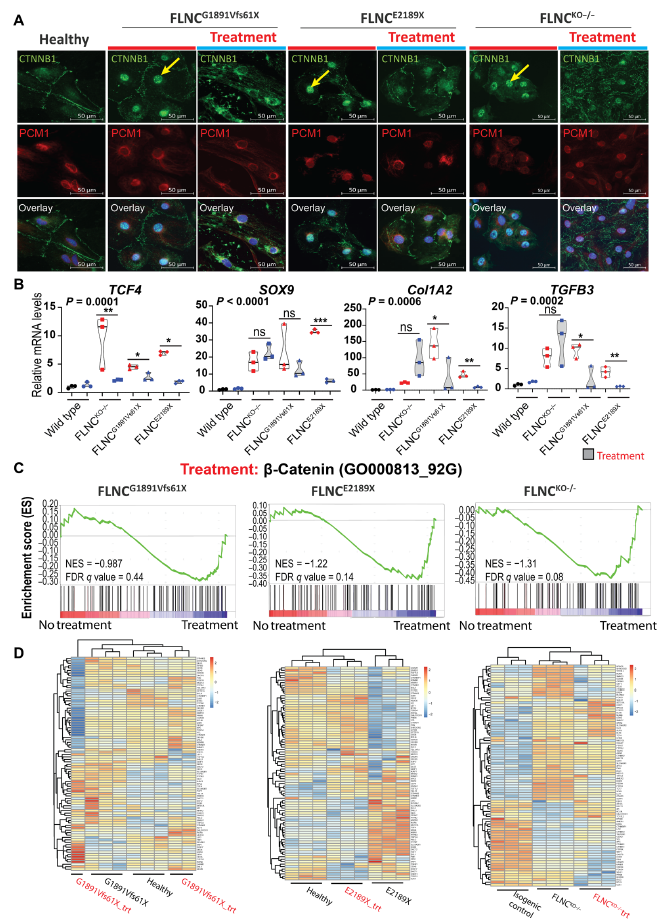
圖7 PDGFRA抑制減緩了β-catenin信號通路及其在FLNC突變體iPSC-CMs中的核定位
7、a-DCM中PDGFRA信號的激活
為了證實體外研究的結果,測定了a-DCM患者心臟中PDGFRA的表達水平。在a-DCM患者中(n = 5), 1例患者是FLNCG1891V61X突變的姐妹。另外4例患者診斷為特發性DCM合并室性心律失常。發現a-DCM患者心臟PDGFRA水平升高, PDGFRA下游效應物ERK被激活(Fig. 8A and B)。此外,還確定了β-catenin和GJA1在DCM心臟中的表達水平和定位。雖然β-catenin和GJA1的表達水平結果保持不變,但免疫熒光染色檢測到β-catenin和GJA1從細胞膜上分離(Fig. 8C and E)。此外,移植心臟的RNA-seq數據顯示,與DCM (n = 15)和未衰竭心臟(n = 12)相比,a-DCM (n = 20)心臟中PDGFRA mRNA的豐度顯著更高(Fig. 8F)。RNA-seq數據經qPCR驗證,如Fig. 8G所示。此外,各組間PDGFRB mRNA轉錄水平未發生變化,提示FLNC突變引起a-DCM的致病機制是特異性于PDGFRA亞型的(Fig. 8F)。總之,證實了來自患者特異性和KO iPSC-CM系和人類a-DCM移植心臟的體外發現,強烈表明PDGFRA促進a-DCM的致病信號傳導。
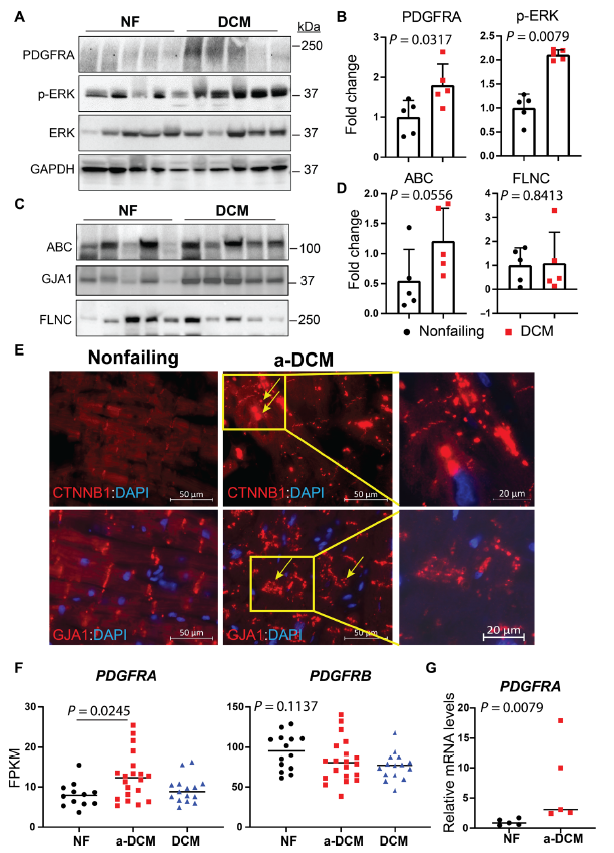
圖8 PDGFRA的上調有助于GJA1和β-catenin在心律失常性擴張型心肌病心臟的離域
總之,如圖9,本研究發現β-catenin是FLNC的下游靶點,FLNC單倍不足不僅影響cell-cell adhesion、細胞骨架和肌肉組織,還增加β-catenin的核遷移,進而上調PDGFRA的轉錄。隨后PDGFRA信號的激活可能是FLNC突變體iPSC-CMs中觀察到的收縮功能障礙和電不穩定的原因。FLNC 單倍不足和隨后PDGFRA激活引起的心律不齊的表現型至少部分是由于GJA1位移引起的細胞膜的幾種機制導致的,如PDGFRA下游靶點ERK磷酸化,ID中斷和異常易位。
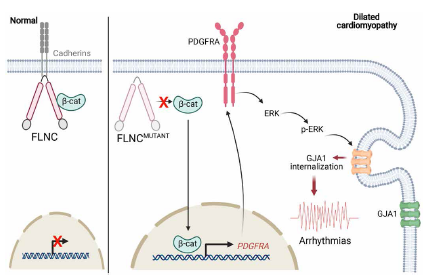
圖9 FLNC相關心肌病的疾病模型
參考文獻:
Chen Suet Nee., Lam Chi Keung., Wan Ying-Wooi., Gao Shanshan., Malak Olfat A., Zhao Shane Rui., Lombardi Raffaella., Ambardekar Amrut V., Bristow Michael R., Cleveland Joseph., Gigli Marta., Sinagra Gianfranco., Graw Sharon., Taylor Matthew R G., Wu Joseph C., Mestroni Luisa.(2022). Activation of PDGFRA signaling contributes to filamin C-related arrhythmogenic cardiomyopathy. Sci Adv, 8(8), eabk0052. doi:10.1126/sciadv.abk0052