






淺談DUSP26誘導主動脈瓣鈣化的機制
鈣化性主動脈瓣疾病(CAVD)的發病率和死亡率仍然很高,而治療選擇有限。在此,作者評估了雙特異性磷酸酶26 (DUSP26)在CAVD中的作用和治療價值。本研究于2021年6月發表在《European Heart Journal》IF:29.983期刊上。
技術路線:

主要實驗結果:
為了鑒定參與CAVD的基因,對人鈣化性主動脈瓣(CAV)進行測序,獲得差異表達基因譜(圖1A)。預實驗中,檢測在CAV組最顯著上調的10個差異表達基因在50對樣本中的表達,有8個和測序結果的趨勢一致,用shRNA干擾他們的表達檢測,發現DUSP26對成骨標志基因的下調最顯著,所以作者將DUSP26用于進一步實驗。圖1B-1D展示了DUSP26的表達,和成骨標志基因的表達。
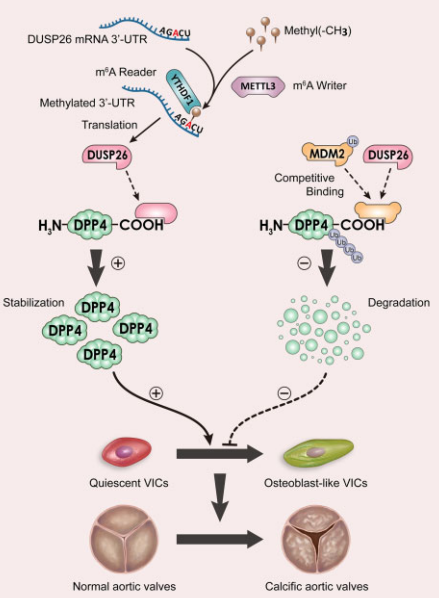
為了評估m6A和DUSP26上調的相關性,作者分析了DUSP26序列,并在其接近終止密碼子的3’-UTR處鑒定到一個METTL3保守序列(AGACU)(圖1E-1F)。成骨刺激增加了原發性hVIC中m6A的整體水平,該作用分別被METTL3的沉默或過表達所減弱和加重(圖1G)。RIP-qRT-PCR(圖1H)和熒光素酶(圖1I)證實了METTL3和DUSP26之間的相互作用。敲除METTL3顯著降低了DUSP26蛋白的表達和半衰期,過表達則相反(圖1J-1M)。這些結果表明METTL3介導的m6A修飾是CAVD中DUSP26表達增加的原因通過增加其穩定性。
METTL3通常以YTHDF1依賴的方式介導m4A修飾,所以探究了YTHDF1的作用。結果顯示敲除YTHDF1顯著降低了DUSP26的蛋白表達和半衰期,過表達則相反(圖1N-1O)。YTHDF1和DUSP26的mRNA序列之間結合,這種結合作用可以被METTL3沉默和過表達所抑制或加強(圖1P-1Q)。

圖1 DUSP26在人鈣化主動脈瓣中上調,并經歷METT3介導的m6A修飾
為了探究DUSP26在體內主動脈瓣鈣化中的作用,作者在ApoE-/-小鼠中敲除DUSP26(AAV2-sh-DUSP26組;AAV2-scr為對照),24周后,HCD t AAV2-scr組最大跨瓣噴射速度和平均跨瓣壓力梯度顯著升高,主動脈瓣面積明顯減少;DUSP26的敲除可以部分回復這些變化(圖2A-2C)。然后檢測了ApoE-/-小鼠瓣葉的形態、纖維化和鈣化。HCD促進主動脈瓣鈣化,表現為主動脈瓣瓣葉厚度增加、膠原增加、鈣離子沉積(圖2D-2G)。而敲除DSP26可以部分恢復上述改變,并降低成骨標志物的表達。
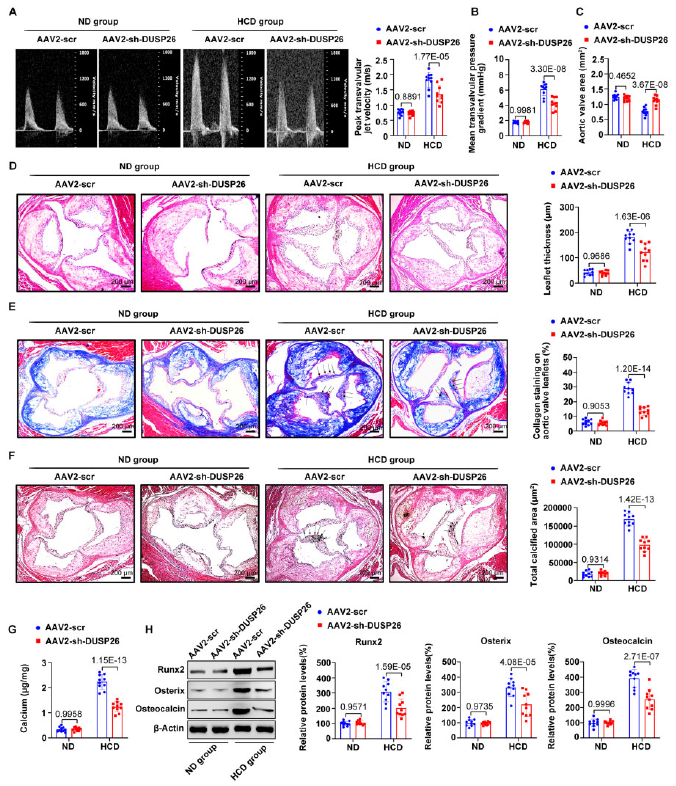
圖2 DUSP26缺失可減輕體內主動脈瓣鈣化
3、DUSP26敲除改善體外主動脈瓣鈣化
作者從人主動脈瓣分離得到hVICs細胞用于體外實驗。用成骨培養基誘導hVICs的成骨分化,發現DSP26的表達隨著分化時間逐漸增加(圖3A-3B)。隨后敲除hVICs中的DUSP26,發現抑制了成骨分化標志基因的表達,以及鈣化結節形成和鈣沉積,過表達DUSP26的效果則相反(圖3C-3D)。這些說明DUSP26沉默在體外也可改善主動脈瓣鈣化。
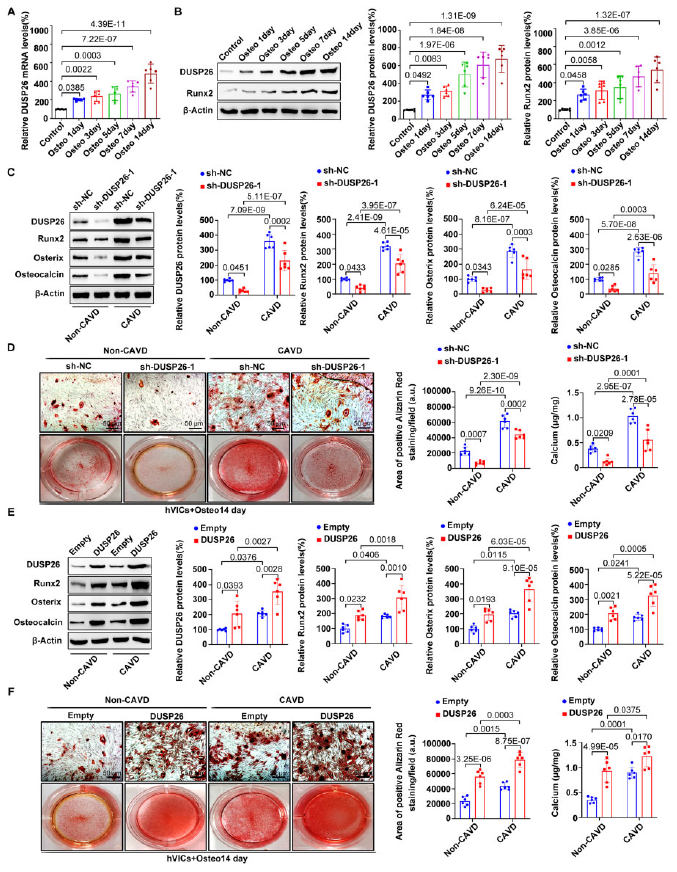
圖3 DUSP26促進hVICs的成骨分化
4、在hVICs中,DUSP26和DPP4相互作用并抑制其降解
首先分離hVICs提取物并進行DUSP26抗體的免疫沉淀,進行MS分析,由此鑒定到DPP4是DUSP26的潛在結合蛋白(圖4A-4B)。Co-IP實驗驗證兩者間的結合關系(圖4C-4D)。DUSP26是一個蛋白磷酸酶,于是探究其酶活性是否是兩者結合所必須的。用F1063-0967處理或突變C152S抑制DUSP26的活性,結果發現其不影響兩者間的結合(圖4E-4F)。因此,DUSP26與DPP4相互作用不依賴于其磷酸酶活性。隨后構建兩個DPP4的截斷性突變體,發現只有C端突變體可以和DUSP26結合(圖4G-4H)。以上說明DUSP26和DPP4的C端結合。
隨后探究DUSP26對DPP4的表達的影響。發現敲除DUSP26后DPP4的蛋白表達顯著下降,過表達則相反,但都不影響mRNA的表達(圖4I-4J)。DUSP26沉默誘導的DPP4下調被26S蛋白酶體抑制劑MG132減弱(圖4K)。此外,DUSP26基因敲除縮短了hVIC中DPP4的半衰期,增加了DPP4的泛素化(圖4L-4M)。

圖4 DUSP26與DPP4相互作用,保護DPP4在hVICs中免受泛素介導的降解
5、DUSP26抑制hVICs中MDM2介導的DPP4的泛素化
作者在DPP4蛋白中發現了一個保守的MDM2結合基序(圖5A)。為了探究MDM2是否作為DPP4的新型E3連接酶,通過co-IP來評估DPP4和MDM2之間的相互作用。在hVIC中異位表達的HA-DPP4與myc標記的MDM2共沉淀(圖5B)。內源性DPP4和MDM2之間的相互作用也證實了(圖5C)。此外,MDM2沉默后增加了DPP4的蛋白水平但對mRNA沒影響(圖5D)。相反,MDM2過表達降低了hVIC中DPP4的蛋白水平,這種作用受到MG132的抑制(圖5E)。此外,MDM2過表達降低了hVIC中DPP4的半衰期(圖5F),并增加了DPP4多聚泛素化(圖5G)。這些數據表明DPP4確實是MDM2的底物。為了確定DPP4與MDM2結合的區域,進行了體外co-IP實驗,發現DPP4的C端與MDM2相互作用(圖5H),之前的結果中,DUSP26也和DPP4的C端作用,所以探索了DUSP26在hVIC中與MDM2結合DPP4競爭的能力。hVIC中DUSP26的沉默增強了MDM2和DPP4之間的相互作用(圖5I)。相反,DUSP26過表達阻斷了MDM2與DPP4的結合(圖5J)。此外,DUSP26敲除降低了DPP4蛋白水平,在hVIC中,這種效應在DUSP26和MDM2共同敲低后減弱(圖5K)。這些結果說明MDM2作為DPP4的新型E3連接酶,DUSP26與MDM2結合在DPP4的C端,從而提高DPP4蛋白在hVIC中的穩定性。
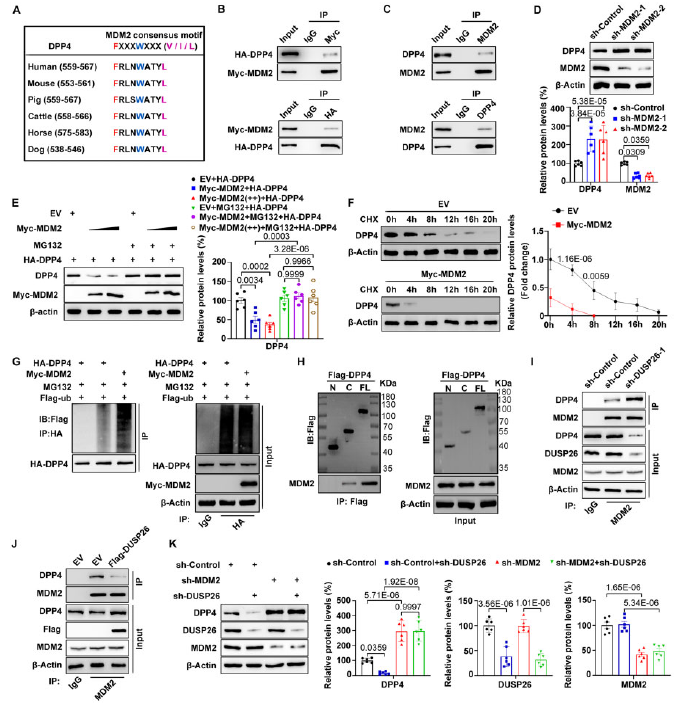
圖5 DUSP26通過在hVIC中與MDM2介導的泛素化競爭來穩定DPP4
6、DPP4過表達挽救了hVIC中DUSP26沉默誘導的成骨分化表型
接下來,進行挽救實驗來確定DUSP26是否通過DPP4的上調來發揮作用。DUSP26敲低的鈣化減緩作用被DPP4過表達所逆轉(圖6A和6B)。相反,DUSP26過表達的促鈣化作用被DPP4過表達所阻止(圖6C和6D),表明DUSP26通過上調DPP4在hVICs中促進成骨分化。
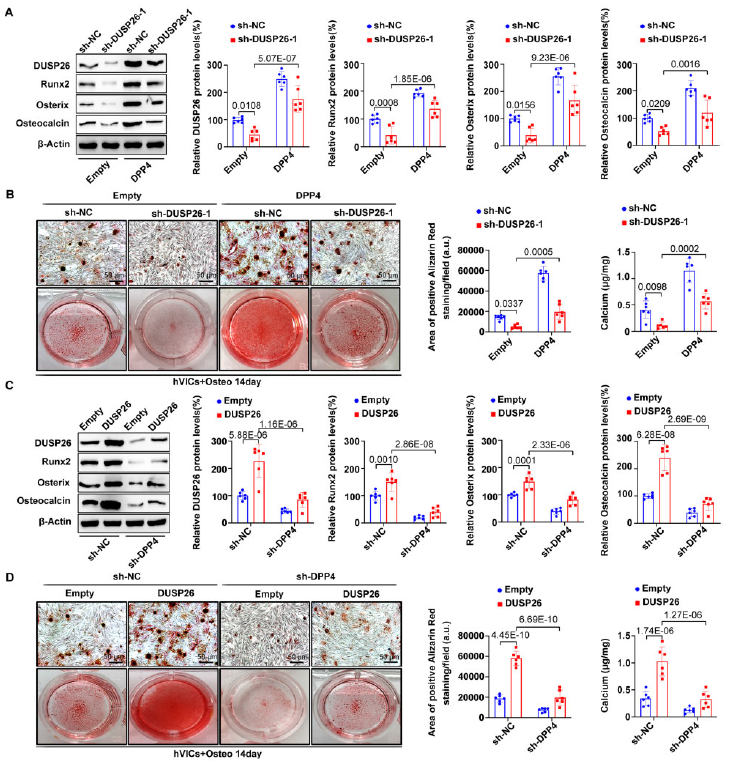
圖6 DPP4過表達挽救了hVIC中DUSP26沉默誘導的成骨分化表型
參考文獻:
Wang Yongjun., Han Dong., Zhou Tingwen., Chen Cheng., Cao Hong., Zhang Joe Z., Ma Ning., Liu Chun., Song Moshi., Shi Jiawei., Jin Xin., Cao Feng., Dong Nianguo.(2021). DUSP26 induces aortic valve calcification by antagonizing MDM2-mediated ubiquitination of DPP4 in human valvular interstitial cells. Eur Heart J, undefined(undefined), undefined. doi:10.1093/eurheartj/ehab316