






LIFR?NF-κB?LCN2軸控制肝臟腫瘤的發生和對鐵死亡的易感性
肝細胞癌(hepatocellular carcinoma,HCC)是肝細胞中最常見的實體腫瘤,嚴重威脅患者的生命健康。鐵死亡在癌癥中具有較大的治療潛力。本研究探討高甲基化水平基因LIFr在調控HCC細胞鐵死亡中發揮的作用。本文于2021年12月發表在《Nature communications》雜志上,IF=12。298。
本研究技術路線:
本研究主要結果:
1. LIFR在HCC中下調,LIFR的缺失促進肝癌的發生
基于TCGA數據庫的測序數據和qPCR分析顯示 ,與正常組織相比,LIFR mRNA水平在HCC中顯著下調 (Fig 1a–c)。接下來作者通過n -亞硝基二乙胺(DEN)誘導小鼠肝臟腫瘤。 5只一歲的DEN處理的C57BL/6小鼠中有4只肝臟腫瘤中Lifr的表達低于正常小鼠肝臟組織 (Fig 1d)。
為了研究LIFR的功能,在 C57BL/6 菌株通過 LoxP-flanked (floxed)處理獲得 Lifr flox/flox(Lifr fl/fl )純合子。然后,作者通過 albumin-Cr小鼠獲得肝細胞特異性Lifr缺失突變體(Lifr fl/ fl; Alb-Cre)小鼠。與預期一樣,Alb-Cre小鼠肝臟中Lifr mRNA和蛋白水平下降 (Fig 1e-f)。為了確定肝臟表型,作者跟蹤了一組小鼠到 2歲。結果發現在14只Lifr fl/ flAlb-Cre小鼠中有4只觀察到肉眼可見的肝臟腫瘤(4只小鼠的腫瘤數量分別為1、2、3和4),而12只Lifr fl/fl小鼠均未顯示可見腫瘤(Fig 1g)。與此同時,14日齡時,作者對Alb-Cre小鼠腹腔注射DEN。并在7個月后對所有小鼠實施安樂死,結果發現在20只Lifr fl/flAlb-Cre小鼠中檢測到4只存在顯著的HCC腫瘤負擔,但在15只Lifr fl/fl對照小鼠中沒有發現 (Fig 1h)。Alb-Cre小鼠從第5個月時開始死于肝臟腫瘤,Lifr fl/fl小鼠從第14個月時開始死于肝臟腫瘤。綜上所述,這些數據表明Lifr是自發和致癌誘導的肝臟腫瘤的抑制因子。此外,LifrLifr的缺失顯著加重了癌基因誘導的肝細胞瘤,并增加了肝臟重量和肝臟體重比(Fig 1j-m)。
為了解成年小鼠中Lifr缺失的影響。枸櫞酸他莫昔芬注射到小鼠中處理5天之后進行觀察,發現Lifr fl/fl ;Cre-ERT2小鼠和Lifr fl/fl小鼠肝癌明顯惡化(Fig 1n-q),表明 LIFR 是成年肝癌基因誘導的抑制因子。
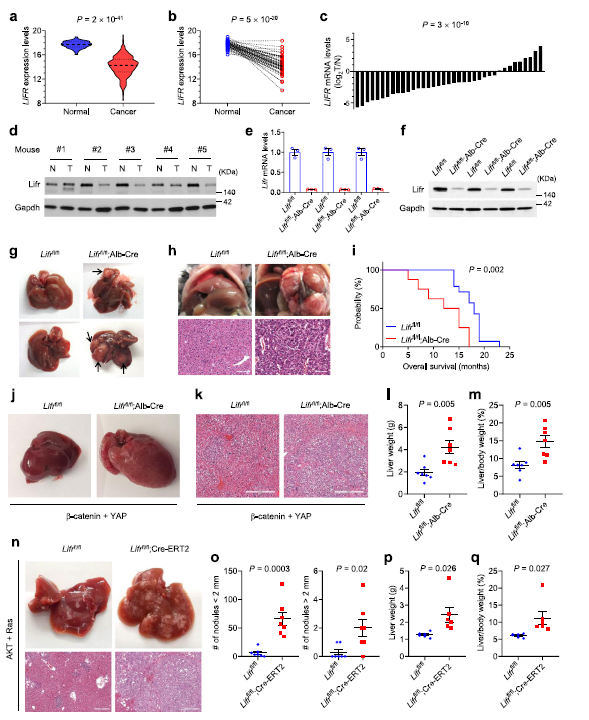
Fig 1 Lifr的缺失會促進肝癌的發生
2. LIFR對誘導鐵死亡的藥物具有敏感性
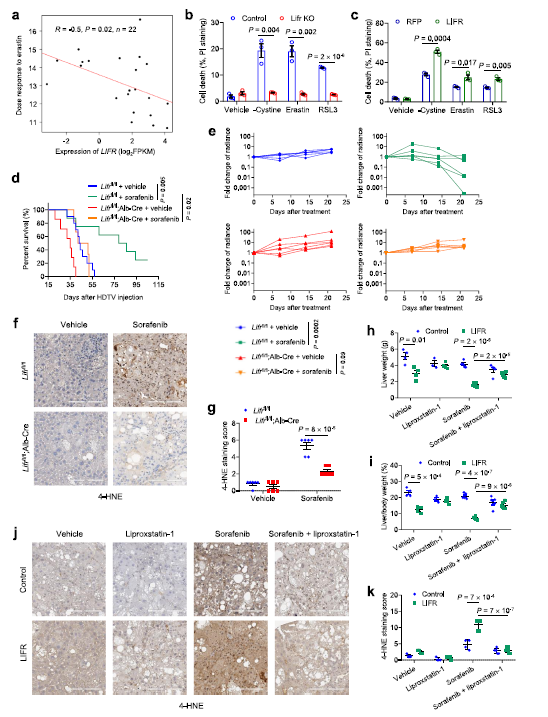
為了評估LIFR的表達是否與藥物反應相關,作者使用了癌癥治療反應門戶(CTRP)分析了基因表達與癌癥細胞系中481種化合物的反應之間的相關性。來自CTRP的肝癌細胞系數據顯示LIFR表達和鐵死亡誘導劑敏感性密切相關 (Fig 2a)。為了確定LIFR是否能調節鐵死亡,作者用鐵死亡誘導物和另外兩種廣泛使用的鐵死亡誘導物RSL3和胱氨酸刺激肝細胞。結果發現這三種處理都觸發了對照組PHM細胞的大量細胞死亡;相比之下,敲除LIFR的PHM細胞對誘導細胞凋亡具有抗性(Fig 2b)。相反,過表達LIFR可使PHM細胞對所有三種鐵誘導物敏感(Fig 2c)。
最近,相關研究報道sorafeni在某些條件下可以觸發鐵死亡。有趣的是,在sorafeni治療后,細胞凋亡或壞死抑制劑(而不是凋亡或壞死抑制劑)可以在體外恢復肝癌細胞活力。這促使作者研究LIFR是否調節索拉非尼的敏感性。對小鼠進行為期一周的sorafeni處理后發現,,索拉非尼治療大大提高了LIFRfl/fl 小鼠存活率(Fig 2d)。索拉非尼治療對LIFRfl/fl 小鼠具有較好的抗腫瘤作用而對LIFRfl/fl Alb-Cre小鼠的作用不明顯(Fig 2e)。
索拉非尼治療后,與LIFRfl/fl Alb-Cre小鼠相比,LIFRfl/fl小鼠中4-HNE 的含量明顯增加(Fig 2 f,g)。索拉非尼治療可使 HCC 腫瘤中4- HNE水平升高(Fig 2j,k) ,而 LIFR 過表達可使4- HNE水平進一步升高,而lipro-1聯合治療可逆轉4-HNE 水平(Fig 2j,k)。這些數據表明 LIFR 抑制HCC 的生長并提高對索拉非尼誘導的鐵死亡敏感性。
3. LIFR是NF-κB信號通路和LCN2的負調控因子
為了確定Lifr丟失對肝臟轉錄組的影響,作者對Lifrfl/fl和Lifrfl/fl;Alb-Cre小鼠肝臟組織進行了RNA-seq分析。Lcn2是Lifrfl/fl;Alb-Cre小鼠肝臟中上調倍數最高的基因之一,已有研究報道Lcn2促進乳腺腫瘤的發生和轉移,其在肝癌中表達上調。由于Lcn2具有胞質和分泌形式,可以由中性粒細胞和肝細胞分泌,于是作者分析了對照細胞和 LIFR 基因敲除后 PHM 細胞分泌的細胞因子,發現 LCN2是 LIFR 喪失后前五位上調的細胞因子之一(Fig 3b)。進一步通過 qPCR (Fig 3 c)和 ELISA (Fig 3 d)驗證了 LCN2在LIFR 敲除 PHM 細胞中的表達,以及 LCN2在LIFRfl/fl、 Alb-Cre 小鼠血清中的表達(Fig 3 e)。LIFR 基因敲除后 PHM 細胞p65磷酸化水平顯著升高,LIFR 基因重新表達可逆轉 p65磷酸化水平(Fig 3f) ,在 LIFR 敲除的肝癌細胞系 Mahlavu 和 PLC/PRF/5中觀察到同樣的效果(Fig 3g -f)。
與lifr缺失的PHM cell相似(Fig 3f),lifr敲除的小鼠肝臟中p65磷酸化(Fig 3i, j)以及Lcn2 mRNA(Fig 3k, l)和蛋白水平均上調(Fig 3m, n),表明NF-κB信號通路的激活。與此相反, Stat3、 Yap、 Akt 和 Erk 的磷酸化水平并沒有顯著下降(Fig 3i,j)。此外,他莫昔芬誘導 LIFR 上調p65磷酸化,但不影響肝內 stat3磷酸化(Fig 3o)。Lcn2在LIFR過表達的PHM細胞中下調,在癌基因誘導的HCC模型中,腺病毒介導的LIFR降低了肝組織中p65磷酸化和Lcn2蛋白水平(Fig 3p, q),并降低了血清中的Lcn2水平。
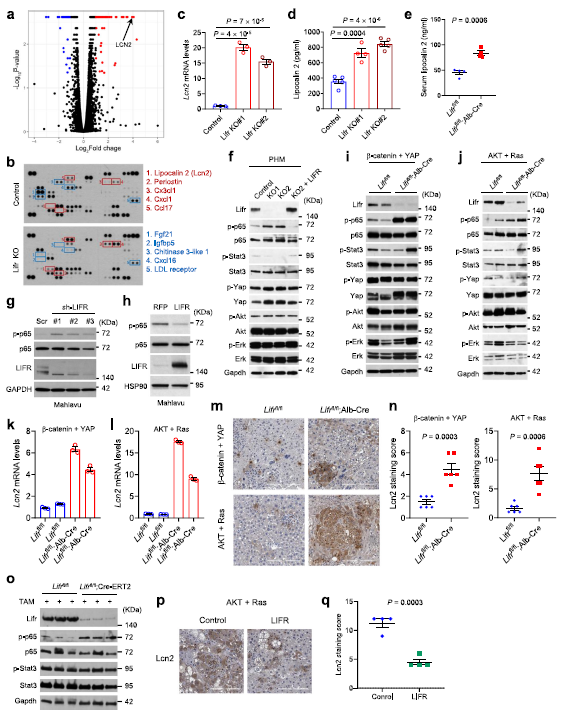
Fig 3 LIFR負向調節NF-κB信號通路和肝臟LCN2
4 . LIFR的缺失通過SHP1激活NF-κB信號通路,導致LCN2的上調
為了了解LIFR如何調節NF-κB信號,作者通過一個蛋白質相互作用數據庫獲取LIFR潛在的相互作用蛋白。在所有候選基因中,磷酸酶SHP1下調肝癌細胞 p65磷酸化已被報道。結果發現,SHP1,而非SHP2,被SFB 標記的LIFR 蛋白通過 s 蛋白珠拉下來,但是沒有被 SFB標記的 GFP 蛋白拉下來(Fig 4 a,b)。SHP1已經被證明可以與TRAF6相互作用,并抑制K63-linked的泛素化,導致NF-κB信號通路失活。而E3連接酶TRAF6介導k63連接的泛素化和TAK1的激酶激活,進而在NF-κB通路中激活IKK。作者過表達LIFR降低了 TRAF6的 k63連鎖泛素化和 p65的磷酸化,這可能是SHP1的降低導致的(Fig 4c),這表明LIFR 通過 SHP1抑制TRAF6的泛素化和 NF-κB 信號轉導。LIFR 在人肝癌細胞系 PLC/PRF/5中的過表達導致 IKKα/β 磷酸化水平下調,IκBα 蛋白水平上調,SHP的降低可消除這些影響(Fig 4d)。此外,LIFR 的降低顯著增加 p65磷酸化,減弱 SHP1和traf6之間的相互作用,而不影響SHP1564位點的磷酸化(SHP1磷酸酶活性的指標)(Fig 4e)。這些數據表明 LIFR 促進SHP1- TRAF6的相互作用,進而抑制 TRAF6泛素化和 NF-κB 信號通路。
為探討NF-κB是否介導LIFR缺失的HEK293T、PLC/PRF/5和PHM細胞株中LCN2的上調,作者使用shRNA敲除了這3個細胞系中的p65,結果發現p65的缺失逆轉了LCN2誘導的LIFR降低(Fig 4f–h)。肝細胞特異性敲除LIFR 顯著增加了肝組織中 LCN2的表達水平,而體內敲除 p65或 LCN2可逆轉這一過程(Fig 4 i,j)。p65 和LCN2的干擾阻斷了肝細胞特異性LIFR基因敲除所誘導的肝癌發生(Fig 4k-m),提示LIFR 基因的缺失一定程度上能通過 NF-κB 和 LCN2促進肝癌的發生。

Fig 4 LIFR的缺失通過SHP1激活NF-κB信號通路,導致LCN2的上調
5. LIFR和SHP1對鐵死亡正向調控,LCN2對鐵死亡負向調控
作者發現,用鐵死亡誘導劑處理HT1080細胞一段時間后會誘導細胞死亡,而敲除LIFR或SHP1可以在鐵死亡誘導劑處理5和10小時后保護HT1080細胞免受細胞死亡 (Fig 5a-d)。此外,LIFR 或 SHP1基因敲除可以減少RSL3、 fin56或 fino2誘導的細胞死亡(Fig 5e,f)。另一方面,隨著時間的推移,LCN2的下調使HT1080細胞敏感,鐵死亡誘導劑誘導的細胞死亡,可以通過與鐵死亡抑制劑liproxstatin-或鐵螯合劑 deoxamine (DFO)的聯合治療來挽救 (Fig 5g)。在用 RSL3,fin56或 fino2處理的ht1080細胞時觀察到類似的效果(Fig 5h)。敲低LIFR 或 SHP1可降低脂質過氧化(Fig 5i,j),而敲低 LCN2可升高脂質過氧化,這中效果能被 liprostatin-1或 DFO 所逆轉(Fig 5k)。這些數據進一步證實 LIFR和 SHP1是鐵死亡的正向調節因子,而 LCN2是鐵死亡的負向調節因子。
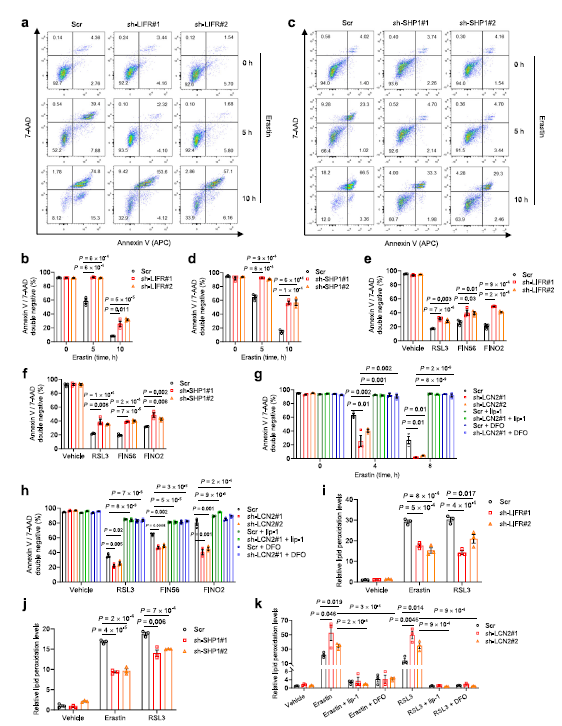
Fig 5 LIF和SHP1對鐵死亡正向調控,LCN2對鐵死亡負向調控
6. LCN2介導鐵死亡耐藥,是提高索拉非尼療效的有效靶點
作者推測 LIFR 通過下調 LCN2調節鐵死亡。實際上,LIFR過表達能提高PLC/PRF/5細胞對鐵死亡誘導劑或索拉非尼的敏感度,這種效果可以通過與 liprostatin-1、 DFO 或純化的 LCN2蛋白協同作用而逆轉,這表明 LIFR 能通過LCN2使肝癌細胞對鐵死亡誘導劑敏感。相反,在 Mahlavu 細胞中敲除LIFR,以及在PHM 細胞中敲除 LIFR,則上調LCN2導致的對鐵死亡誘導劑和索拉非尼耐藥性,而這種耐藥性可以通過敲低LCN2而逆轉(Fig 6 a,b)。
肝臟中LIFR缺失會引起鐵(Fe2 +)水平下調(Fig 6c)。作者假設 lcn2的缺失是通過提高細胞中 Fe2 + 和脂質過氧化的水平來增加對鐵死亡的敏感性。LIFR的缺失降低了細胞內 Fe2 + 水平和脂質過氧化水平,而 lcn2的敲除可以逆轉這兩種效應(Fig 6d,e)。另一方面,LIFR或 lcn2的缺失并沒有改變谷胱甘肽(GSH)、 Slc7a11、 fsp1和 gpx4的水平(Fig 6f,g)。作者測量了LIFR敲除的PHM小鼠肝細胞系中鐵的含量,結果發現LIFR的缺失降低了細胞中Fe2+和脂質過氧化的水平(Fig 6 d,e)。另一方面,LIFR或Lcn2的缺失并沒有改變谷胱甘肽(GSH),Slc7a11, Fsp1和Gpx4的水平 (Fig 6f,g)。
為了進一步探索LCN2靶向制劑是否能提高索拉非尼的治療效果,作者在 NSG小鼠中建立了4個 HCC 異種移植(PDX)模型。結果發現四個 PDX 株系(# 3-# 6)中有三個表現為 LIFR低表達,同時 LCN2和磷酸化 p65水平較高(Fig 6h)。在所有品系中,其中# 5株系LIFR 水平最低,lcn2水平最高,# 4株系則相反(Fig 6h)。在臨床實驗中,將#4和# 5 PDX中的PDX腫瘤組織植入NSG小鼠,當腫瘤達到50 – 100 mm3時開始治療。將小鼠分為4個治療組:(1)IgG + vehicle;(2) LCN2抗體+ vehicle;(3) IgG +索拉非尼;(4) LCN2抗體+索拉非尼。結果發現LCN2抗體+ vehicle以及LCN2抗體+索拉非尼組LCN2水平明顯降低(Fig 6i)。無論是否與索拉非尼聯合治療,PDX # 4的小鼠中,LCN2聯合抗體治療不改變腫瘤的生長(Fig 6j,l)。在PDX # 5的小鼠中,單獨使用LCN2聯合抗體治療對腫瘤生長的影響不大,而聯合治療比單獨使用索拉非尼治療具有更好的抗腫瘤效果(Fig 6k,l)。抗LCN2治療并不影響增殖Ki-67和凋亡標記物caspase3的水平,而與索拉非尼聯合治療提高了4-HNE和 MDA(脂質過氧化的兩個標記物)的水平(Fig 7a,b)。此外,電鏡分析顯示,在 PDX # 5,聯合治療組的腫瘤細胞中含有萎縮的線粒體和嚴重濃縮的膜,這是鐵死亡典型的形態學特征。索拉非尼或LCN2抗體單獨治療也能增加線粒體膜密度,但程度要小于它們聯合治療(Fig 7c)。在 PDX # 4中,無論是單獨使用索拉非尼治療還是聯合治療都能誘導磷酸化鐵蛋白相關的線粒體形態改變,而抗lcn2治療并不能改變線粒體形態(Fig 7d)。總之,一種中和LCN2的抗體增強了索拉非尼對低LIFR表達和高LCN2表達的肝癌患者來源的異種移植瘤的鐵誘導作用。
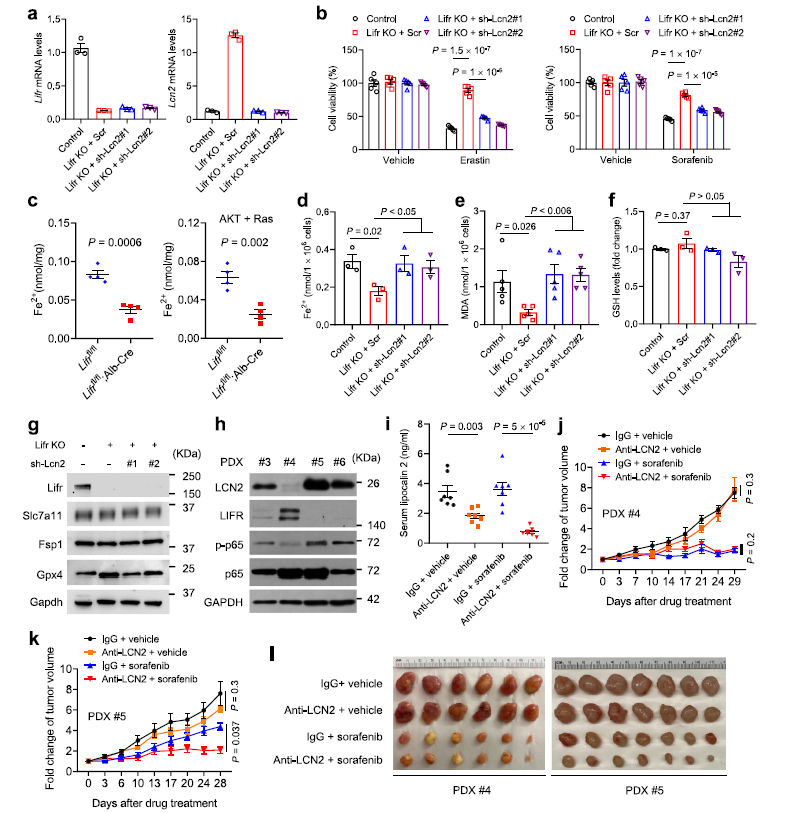
Fig 6 LCN2介導鐵死亡耐藥,是提高索拉非尼療效的治療靶點
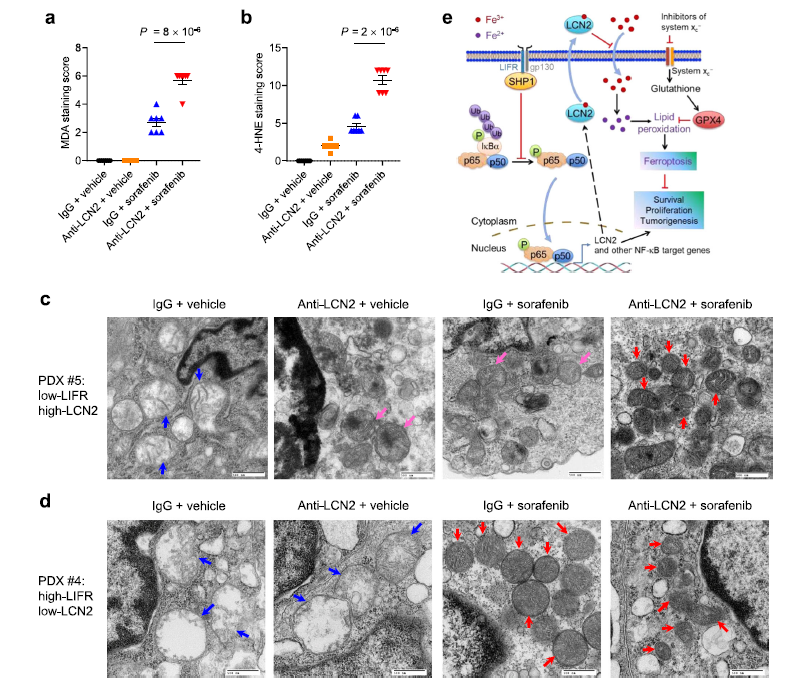
Fig7一種中和LCN2的抗體增強了索拉非尼對低LIFR表達和高LCN2表達的肝癌患者來源的異種移植瘤的鐵誘導作用。
LIFR 的缺失通過 SHP1 激活NF-κB 信號傳導,導致鐵螯合細胞因子 LCN2 的上調,導致對對鐵死亡誘導劑不敏感。因此抗 LCN2 療法可以通過靶向鐵死亡來改善肝癌治療。
參考文獻:
[1] Yao Fan,Deng Yalan,Zhao Yang et al。 A targetable LIFR-NF-κB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis。[J] 。Nat Commun, 2021, 12: 7333。