






HIF-2α激活通過增加細胞內的鐵來增強結腸癌細胞的氧化死亡

結腸癌(CRC)是世界上第三常見的癌癥,也是癌癥相關死亡的主要原因。癌細胞迅速擴張,所有實體瘤由于血管化不足而經歷缺氧。缺氧是實體腫瘤的一個標志,它促進細胞生長、生存和轉移,并對化療和放療產生耐藥性。缺氧反應主要由轉錄因子缺氧誘導因子1α (HIF-1α)和HIF-2α介導,而HIF-1α和HIF-2α在CRC中表現出不同的作用。我們的研究表明,癌癥細胞依賴于HIF-2α的一種機制脆弱性,可用于CRC治療。該研究2021年6月發表在《JOURNAL OF CLINICAL INVESTIGATION》,IF為14.808。
技術路線:

主要研究結果:
1. 藥物篩選確定腫瘤類腸道中HIF-2α的合成易損性
作者構建Apc缺失型(Cdx2-ERT2Cre;Apcfl/fl)和CRC HIF-2α過表達小鼠模型(Cdx2-ERT2Cre;Apcfl/fl HIF-2αLSL/LSL),從這2個小鼠模型中分離腸類藥物,并用化療藥物培養,生長被監測了5天(圖1A)。在Cdx2-ERT2Cre;在Apcfl/fl HIF-2αLSL/LSL小鼠中,他莫昔芬可誘導HIF-2α,并特異性地破壞結腸上皮細胞中的Apc。來自Cdx2-ERT2Cre;Apcfl/fl腫瘤腸系膜的小鼠對doxorubicin, mitoxantrone, irinotecan,以及 eribulin等藥物高度敏感,與來自Cdx2-ERT2Cre;Apcfl/fl HIF2αLSL/LSL的不同(圖1B)。RSL3、sorafenib索拉菲尼、erastin和DMF被認為是最有效的小分子,可以顯著降低Cdx2-ERT2Cre;Apcfl/fl HIF2αLSL/LSL腫瘤腸道類物質的生長(圖1C)。Erastin和RSL3是經典的ferroptosis激活劑,分別抑制xCT(由Slc7a11基因編碼,是xC系統的一個組成部分)和GPX4。DMF一種細胞可滲透的線粒體衍生物,在一些癌癥細胞系中具有細胞毒性(圖1D)這些結果表明,HIF-2α表達的腫瘤可以被氧化應激激活劑選擇性靶向。
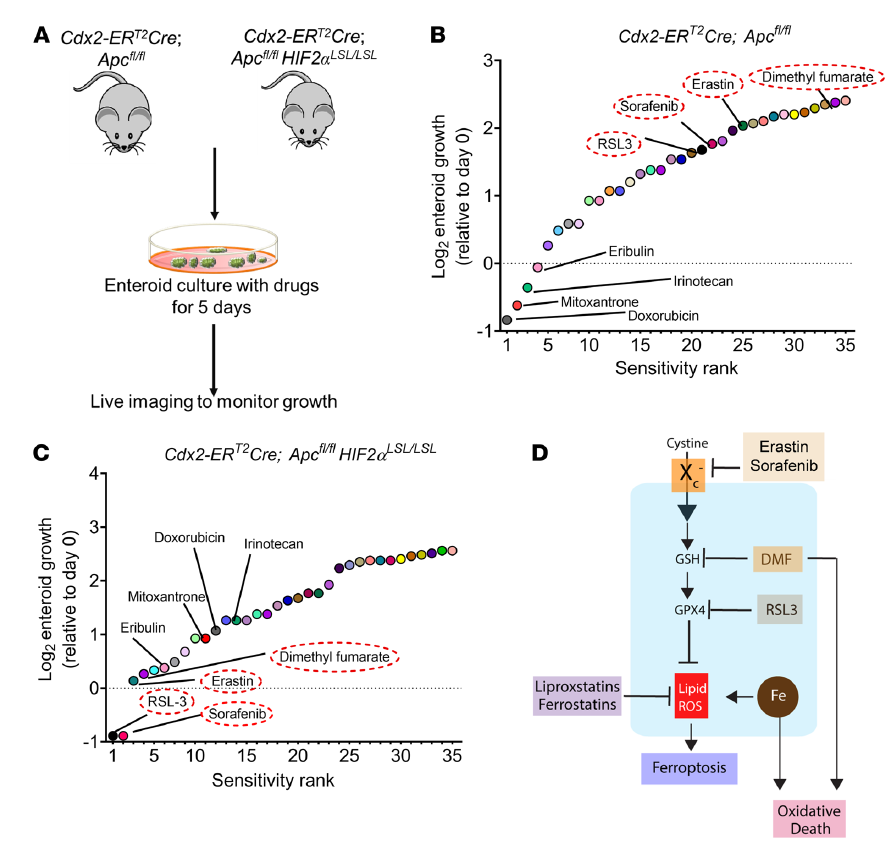
圖1 篩選能抑制過表達HIF-2α的腫瘤腸樣細胞生長的化合物
Erastin和RSL3是經典的鐵死亡誘導劑,作者已證明,缺氧模擬FG4592或缺氧顯著增強了鐵死亡誘導物erastin和RSL3以HIF-2α依賴的方式處理后的細胞死亡。作者在最后一次給藥后14天,分析這些小鼠的結腸組織的組織學變化(圖2A)。Slc7a11的缺失或HIF-2α的過度表達與對照動物的情況無法區分(圖2B)。然而,Slc7a11的破壞與HIF-2α過表達聯合導致結腸上皮變性和空泡化(圖2B)。通過4-羥基2-壬烯醛(4-HNE)染色測定脂質過氧化誘導的氧化應激(圖2C)。與對照組相比,Villin-CreERT2Slc7a11fl / fl; HIF-2αLSL/LSL小鼠的組織學評分(圖2D)和4-HNE強度(圖2E)均顯著增加,表明這些小鼠氧化應激和上皮細胞損失增加。此外,與對照組比較,Villin-CreERT2-HIF-2αLSL/LSL小鼠的HILPDA和PLIN2 mRNA水平顯著升高(圖2F)。與對照組相比,Villin-CreERT2-HIF-2αLSL/LSL小鼠肝臟和腸道鐵水平都更高(圖2G)。肝臟鐵含量的增加是由于小腸對鐵的吸收增加。這些數據證實了HIF-2α在體內對鐵死亡敏感的作用,并提示誘導鐵死亡的藥物可能對缺氧細胞有很高的殺傷效果。

圖2. HIF-2α激活可增強體內鐵死亡
3. HIF激活可促進DMF誘導的CRC細胞死亡
作者的篩選鑒定出DMF是一種小分子,可以有效地減少低氧腫瘤腸道樣體的生長(圖1C)。一組CRC細胞系使用DMF單獨或聯合缺氧或模擬缺氧FG4592處理。通過MTT和長期克隆生存試驗評估,FG4592增強DMF誘導的CRC細胞死亡(圖3,A- C)。此外,DMF處理和缺氧培養的細胞存活率較低(圖3,D和E)。與erastin和RSL3數據一致,HIF-2α是促進DMF介導的CRC細胞生長的關鍵(圖3F)。

圖3. 缺氧模擬有助于DMF誘導的CRC細胞生長抑制
4. DMF獨立于鐵死亡誘導細胞死亡
由于DMF與其他細胞對鐵死亡激活劑一起有效地降低了缺氧細胞的生長(圖1C),我們評估了DMF是否介導了鐵死亡激活劑的死亡。在DMF處理后,Fer-1和Lip-1不能挽救細胞的死亡和活力,而rsl3介導的細胞死亡被Fer-1和Lip-1挽救(圖4A)。在DMF處理后,Fer-1和Lip-1不能挽救細胞的死亡和活力,而RSL3介導的細胞死亡被Fer-1和Lip-1挽救(圖4A)。DMF可與抗氧化劑GSH直接反應,導致NADPH降低,ROS增強。然而,FG4592和糖酵解抑制劑的共同作用并沒有降低細胞活力(圖4E)。這些結果表明,在DMF介導的缺氧腫瘤細胞死亡中有其他機制參與。
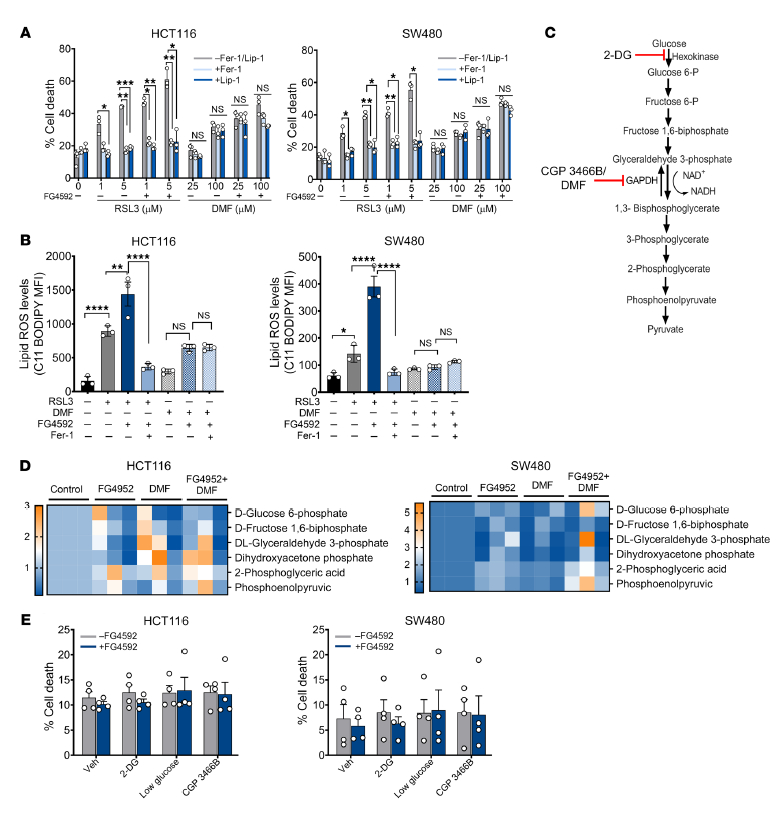
圖4. DMF在CRC細胞中不是鐵轉移誘導物。
5. 在DMF誘導的HIF-2α介導的細胞死亡中,活性氧積累和鐵毒性是必不可少的
添加半胱氨酸前體n -乙酰半胱氨酸(NAC)的生長培養基挽救了含或不含FG4592 DMF處理的HCT116和SW480細胞的死亡和活力(圖5A和補充圖9C)。在DMF處理和維持缺氧的細胞中也觀察到類似的結果(圖5B)。FG4592或單獨的缺氧處理增加了ROS,這是通過細胞滲透的2,7 -二氯二氫熒光素二醋酸酯(H2DCFDA)評估的,它被用作ROS的指標(圖5,C和D)。與DMF的協同處理增強了ROS生成的增加,而NAC可以挽救ROS生成(圖5,C和D)。為了證實對氧化細胞死亡的敏感性是由HIF-2α介導的,利用了shRNA介導的HIF-1α和HIF-2α敲低細胞。HIF-2α敲低細胞中的ROS水平顯著降低(圖5E),表明DMF處理后HIF-2α在ROS產生中的作用。
由于HIF激活會導致線粒體代謝的變化和ROS的產生,作者分析了DMF或FG4592處理是否會引起線粒體代謝物池的變化。然而,線粒體代謝物水平未見顯著變化,表明HIF-2α通過其他機制影響細胞ROS(圖5F)。由DMF和FG4592介導的細胞死亡在低鐵和對照培養基中被挽救(圖5G)。總之,這些數據表明,鐵毒性通過HIF-2α和氧化應激脆弱性的機制。
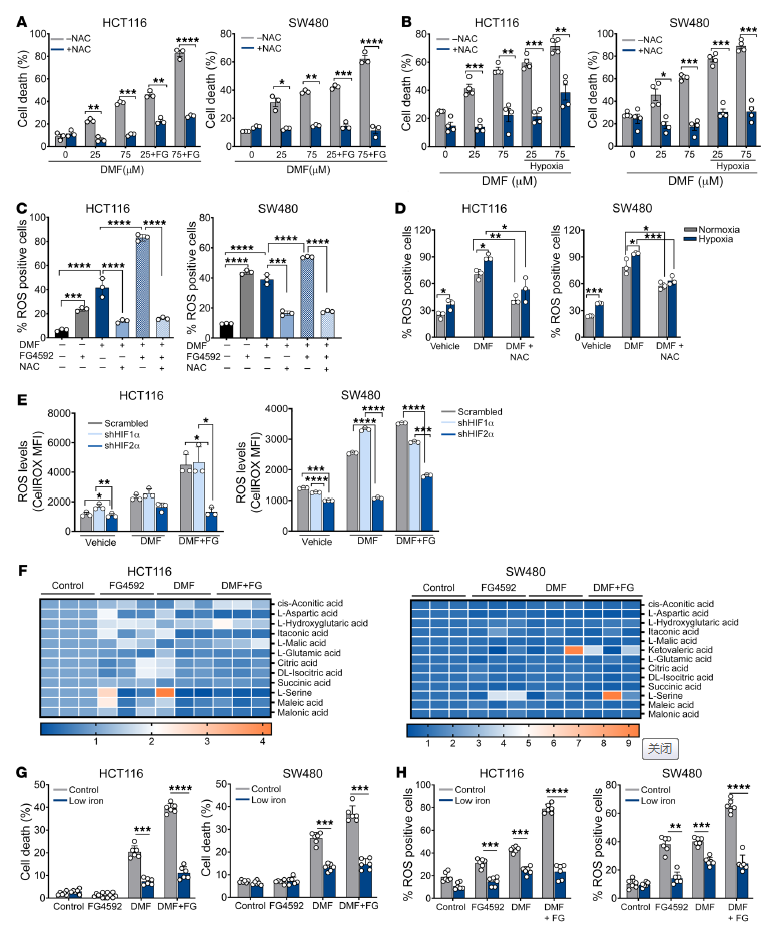
圖5. 活性氧的產生和鐵的積累參與DMF和FG4592介導的CRC細胞死亡
6. H2S的保護性過硫化作用可以挽救DMF和HIF-2α誘導的細胞死亡
接下來,評估了不可逆的蛋白質氧化是否參與了DMF和FG4592誘導的細胞死亡。H2S可通過過硫化作用(翻譯后修飾)防止半胱氨酸硫醇的過度氧化(圖6A) 為了排除H2S的保護作用不是由于DMF上的硫化物陰離子親核添加導致的DMF消耗,在DMF和FG4592或DMF和缺氧導致細胞死亡16小時后,將Na2S和3-MP添加到新鮮培養基中(圖6B)。即使在這些條件下,Na2S和3-MP也能防止細胞死亡(圖6,C和D)。此外,補充3-MP和Na2S后,DMF或DMF和FG4592誘導的細胞內ROS水平降低(圖6E)。這些數據與模型一致,即DMF增強HIF誘導的細胞死亡涉及氧化蛋白損傷,而氧化蛋白損傷受到H2S的保護。

圖6. H2S可防止不可逆的蛋白質氧化,挽救DMF和FG4592介導的細胞死亡
7. FG4592增強DMF介導的體內CRC細胞死亡
接下來,作者評估了DMF和FG4592聯合治療已建立的CRC腫瘤的體內療效。此,HCT116、SW480和DLD1細胞被皮下植入免疫缺陷小鼠的側壁,并在DMF和FG4592治療前建立10天(圖7A)。從與接受對照或單獨藥物治療的患者相比,在所有3個異種移植中,DMF和FG4592治療的腫瘤體積(圖7B)和腫瘤重量(圖7C)均顯著減少。通過BrdU摻入試驗評估的腫瘤細胞增殖在DMF和FG4592聯合處理的HCT116、SW480和DLD1異種移植中也降低了(圖7D)。然而,在與DMF和FG4592共同處理的所有3個異種移植物中,tunel陽性凋亡細胞的百分比都有所增加(圖7E)。總之,這些數據表明,缺氧腫瘤細胞對DMF治療非常脆弱,突出了一個潛在的治療窗口。
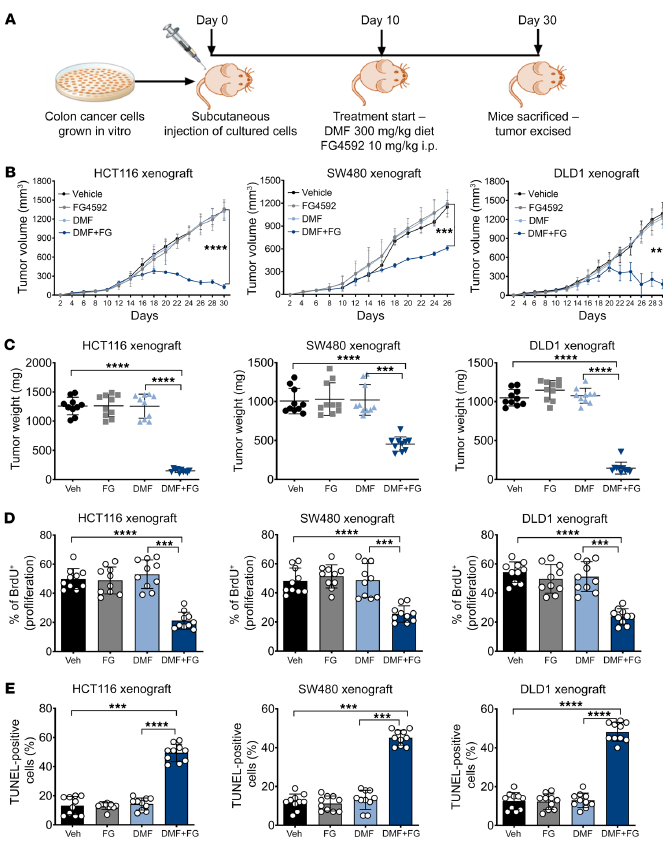
圖7. DMF和FG4592增強體內CRC細胞死亡
為了證實HIF-2α在DMF介導的體內CRC細胞死亡中的作用,利用HIF-2α敲低HCT116細胞。在DMF和FG4592治療前,將穩定的非靶標擾亂和HIF-2α敲低的HCT116細胞皮下注射到免疫受損小鼠的兩側,并允許其生長10天(圖8A)。HIF-2α基因敲低的細胞對DMF和FG4592處理具有抗性。在DMF+FG4592處理的小鼠中,表達雜亂shRNA的細胞顯示腫瘤體積和重量顯著減少,而HIF-2α敲低的細胞則完全耐受(圖8,B和C)。同樣,在DMF+FG4592處理后,HIF-2α敲低細胞的腫瘤增殖和凋亡沒有改變(圖8,D和E)。這些數據表明,HIF-2α激活增加了體內氧化細胞死亡的脆弱性。
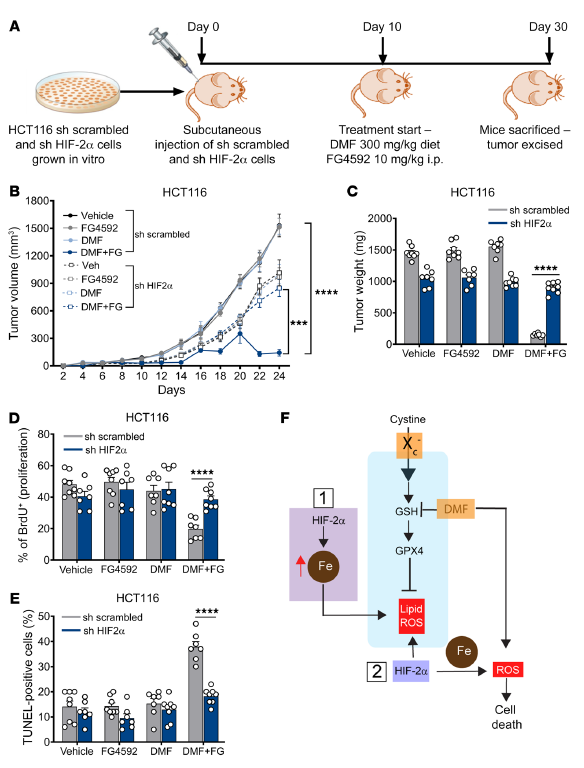
圖8. DMF介導的體內CRC細胞死亡依賴于HIF-2α
總結:
該研究揭示了HIF-2α在驅動低氧CRC細胞對氧化應激誘導化合物(如DMF)的合成致命性方面的作用,并揭示了利用這種內在脆弱性進行化療發展的潛力。