乳腺癌衍生的外泌體 miR-138-5p 通過抑制 KDM6B 調節腫瘤相關巨噬細胞的極化
乳腺癌是女性腫瘤中最普遍的惡性腫瘤,近些年乳腺癌發病率和死亡率逐年增加。乳腺癌微環境不僅包括腫瘤細胞,還包括基質細胞和不同的免疫細胞亞群。其中腫瘤相關巨噬細胞 (TAM) 是腫瘤微環境的關鍵組成部分,廣泛參與調節腫瘤進展。今天我們來講一篇關于乳腺癌外泌體miRNA介導TAM極化的文章,文章題名為:Cancer-derived exosomal miR-138-5p modulates polarization of tumor-associated macrophages through inhibition of KDM6B,發表于Theranostics期刊(IF=11.55)。
miR-138-5p抑制巨噬細胞表達KDM6B
首先檢查了 MB-MDA-231 乳腺癌細胞對巨噬細胞樣細胞系 THP-1 中KDM6B表達的影響。與 MDA-MB-231 懸浮共培養 48 小時后,THP-1 細胞中的 KDM6B 水平顯著降低,源自 MDA-MB-231 的條件培養基 (CM) 抑制了 THP-1 細胞中KDM6B 的表達,表明乳腺癌細胞分泌的一種因子是造成這種效應的原因。
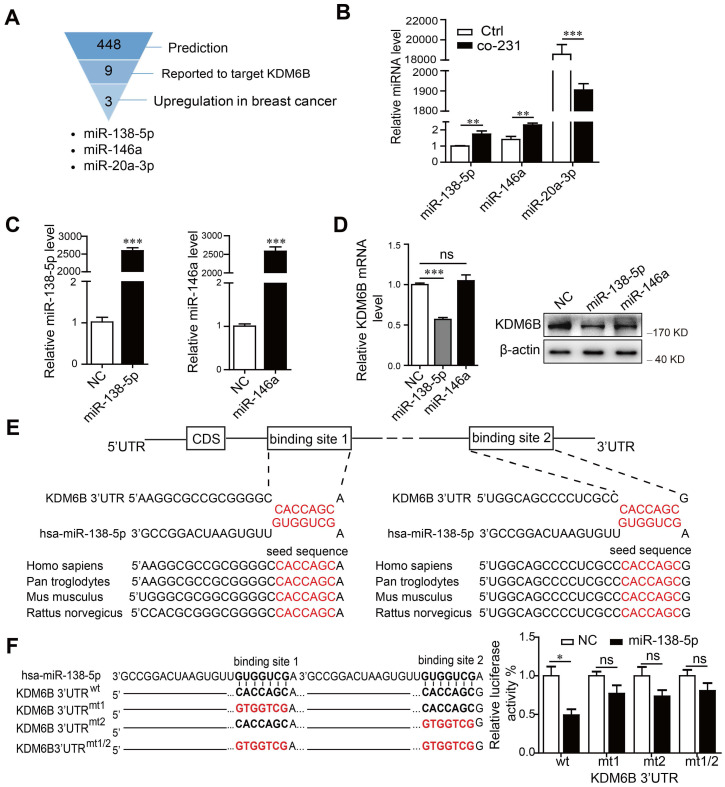
miRNA 參與填充腫瘤微環境的不同細胞類型之間的信號轉導。因此,試圖確定 miRNA 是否有助于KDM6B表達的調節。為此進行了生物信息學分析和文獻綜述,確定靶向KDM6B 3'-UTR的miRNA。確定了miR-138-5p、miR-146a和miR-20a-3p作為候選miRNA。當我們分析與MDA-MB-231細胞共培養后 THP-1 細胞中 miRNA 水平的變化時,發現miR-138-5p和miR-146a顯著增加,而miR-20a-3p減少。考慮到 miRNA 水平通常與靶基因的水平呈負相關,接下來確認了miR-138-5p或miR-146a是否抑制了KDM6B表達。結果發現是miR-138-5p而非miR-146a的異位表達抑制了THP-1細胞KDM6B的表達。序列分析表明,miR-138-5p 在KDM6B mRNA的 3-'UTR 中含有兩個潛在的靶位點。此外,KDM6B mRNA 3-'UTR 中 miR-138-5p 的靶結合序列在眾多不同物種中高度保守。熒光素酶報告基因檢測表明,miR-138-5p 的過表達抑制了KDM6B mRNA的 3'-UTR 驅動的熒光素酶活性,但不會抑制突變的同源 3'-UTR。這些數據表明 miR-138-5p通過與其3'-UTR 結合來抑制KDM6B表達。
miR-138-5p介導癌細胞誘導的巨噬細胞KDM6B表達抑制
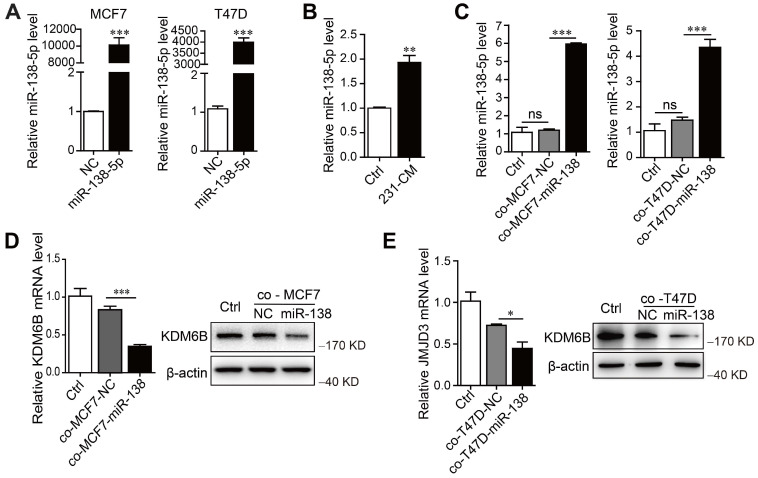
首先測量了表型不同的乳腺癌細胞系以及非致瘤細胞系中的 miR-138-5p 水平。當miR-138-5p mimics轉染 MCF7 和 T47D 細胞過表達 miR-138-5p 時,發現用MDA-MB-231細胞的 CM 處理或與過表達 miR-138-5p的MCF7和 T47D細胞共培養增加了 THP-1 細胞表達的 miR-138-5p 水平。此外,與過表達 miR-138-5p的MCF7和T47D細胞共培養抑制了THP-1細胞中KDM6B mRNA 和蛋白質的表達。因此,得出結論,miR-138-5p 介導了THP-1 細胞與癌細胞共培養誘導的KDM6B表達抑制。
癌細胞產生的外泌體miR-138-5p下調巨噬細胞中KDM6B的表達
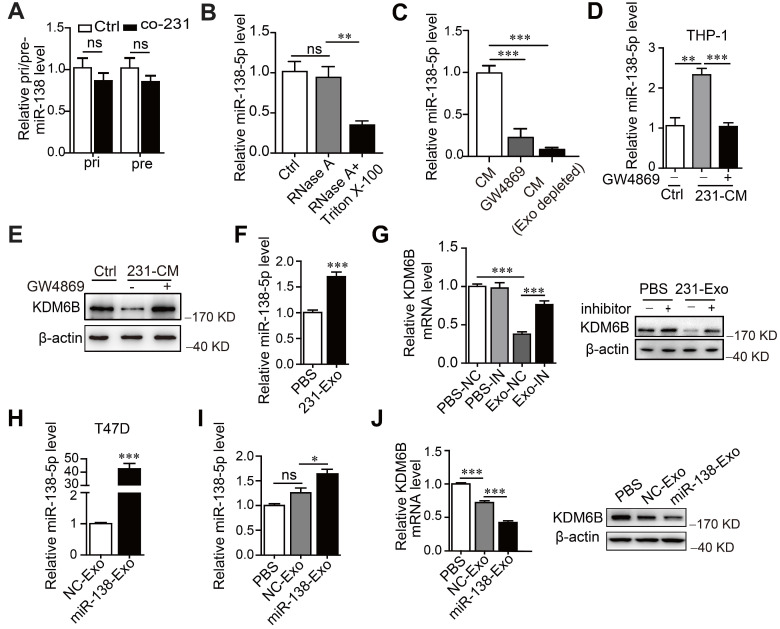
接下來探索miR-138-5p的水平如何增加。為此測量了巨噬細胞中初級 (pri-) 和前體 (pre-) miR-138 的水平。發現處理后THP-1細胞中pri-或pre-miR-138沒有增加,表明miR-138-5p是外源供應的。此外,與用 RNase A 加 Triton X-100 處理相比,當我們用 RNase A 處理 MDA-MB-23 的 CM 時,miR-138-5p的水平沒有變化。這些發現支持細胞外miR-138-5p被包裹在膜結構中的結論。通過觀察到與未處理的CM相比,GW4869 處理的CM或外泌體耗盡的 CM 中miR-138-5p的水平顯著降低,獲得了進一步的證據,表明外泌體miR-138-5p參與了KDM6B表達的調節。這些結果表明 miR-138-5p 被包裹在乳腺癌細胞釋放的外泌體中。
接下來研究了來自乳腺癌細胞的外泌體miR-138-5p是否介導了THP-1細胞中miR-138-5p水平的增加。發現外泌體抑制劑GW4869阻斷了由MDA-MB-231 細胞的CM處理引起的miR-138-5p水平的增加。此外,在THP-1細胞中檢測到 Cy3標記的miR-138-5p,并與Dio標記的外泌體膜共定位。總之,數據表明癌細胞分泌外泌體miR-138-5p被轉移到巨噬細胞。
當測量外泌體miR-138-5p對巨噬細胞中KDM6B表達的影響時,發現外泌體的抑制阻斷了通過用 CM 處理細胞誘導的KDM6B抑制。MDA-MB-231衍生的外泌體的處理增加了THP-1細胞miR-138-5p的水平并抑制KDM6B的表達,這種作用在用miR-138-5p inhibitor處理的THP-1細胞中被消除。用miR-138-5p mimics轉染T47D細胞會增加外泌體miR-138-5p的水平。來自過表達miR-138-5p的T47D細胞的外泌體增加了miR-138-5p水平并抑制了THP-1細胞中KDM6B 的表達。總之,這些結果表明癌細胞分泌的外泌體 miR-138-5p 被遞送到巨噬細胞,在其中它抑制KDM6B。
外泌體 miR-138-5p 通過抑制KDM6B表達調節巨噬細胞極化
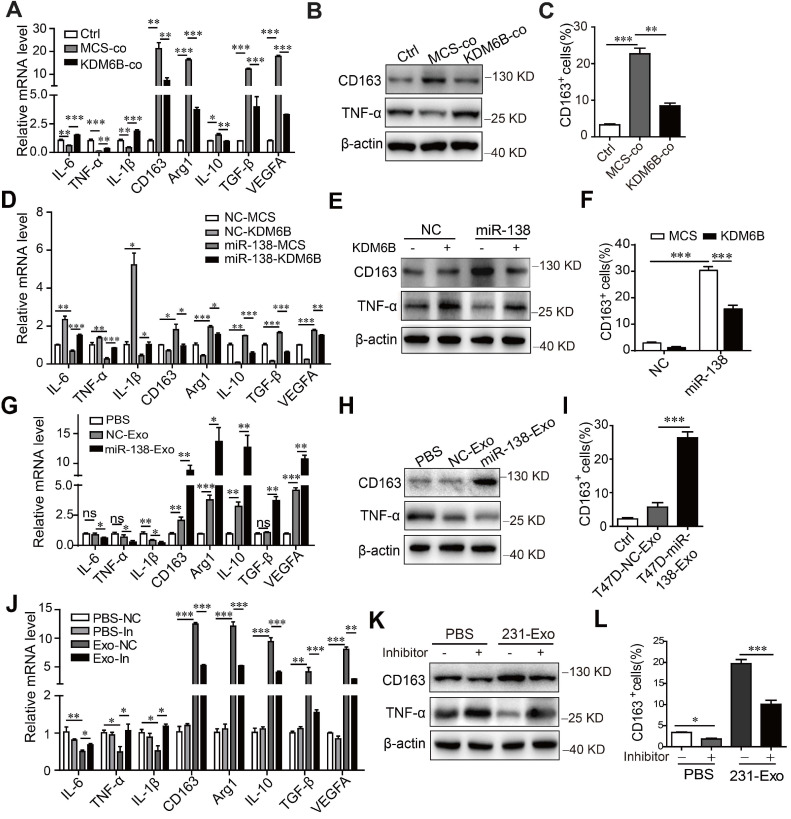
為了確定miR-138-5p 誘導的KDM6B表達抑制是否影響巨噬細胞極化,在不同條件下培養的THP-1細胞的轉錄譜。首先,與腫瘤細胞共培養抑制了編碼 IL-6、TNF-α和IL-1β的M1相關基因的表達,并增加了編碼 CD163、Arg1、IL-10、TGF-β 和M2相關基因的表達。流式細胞術顯示,MDA-MB-231細胞與THP-1細胞共培養增加了CD163陽性巨噬細胞的百分比,當這些細胞過表達KDM6B 時,CD163 陽性巨噬細胞的百分比被抑制。類似地,KDM6B 過表達抑制了用 miR-138-5p mimics轉染THP-1細胞誘導的 M2 樣極化。與對照相比,用從表達 miR-138-5p 的T47D細胞中分離的外泌體處理THP-1細胞降低了M1相關表型。相比之下,巨噬細胞表達的 M2 標志物隨著巨噬細胞培養物中 CD163 陽性細胞數量的增加而增加。
用從 MDA-MB-231 細胞中分離的外泌體處理 THP-1 細胞導致 M2 樣極化,這通過抑制 miR-138-5p 表達來挽救。此外,用 miR-138-5p mimics轉染或與過表達 miR-138-5p的T47D 細胞的 CM 中分離的外泌體孵育增加了THP-1 細胞的增殖。Transwell 分析顯示,用來自MDA-MB-231 CM外泌體 miR-138-5p 孵育的巨噬細胞處理后,細胞的遷移增加。總之,數據表明外泌體miR-138-5p 通過抑制巨噬細胞中KDM6B 的表達來促進 M2 樣極化。
KDM6B 去甲基化酶活性促進 M1 相關基因表達
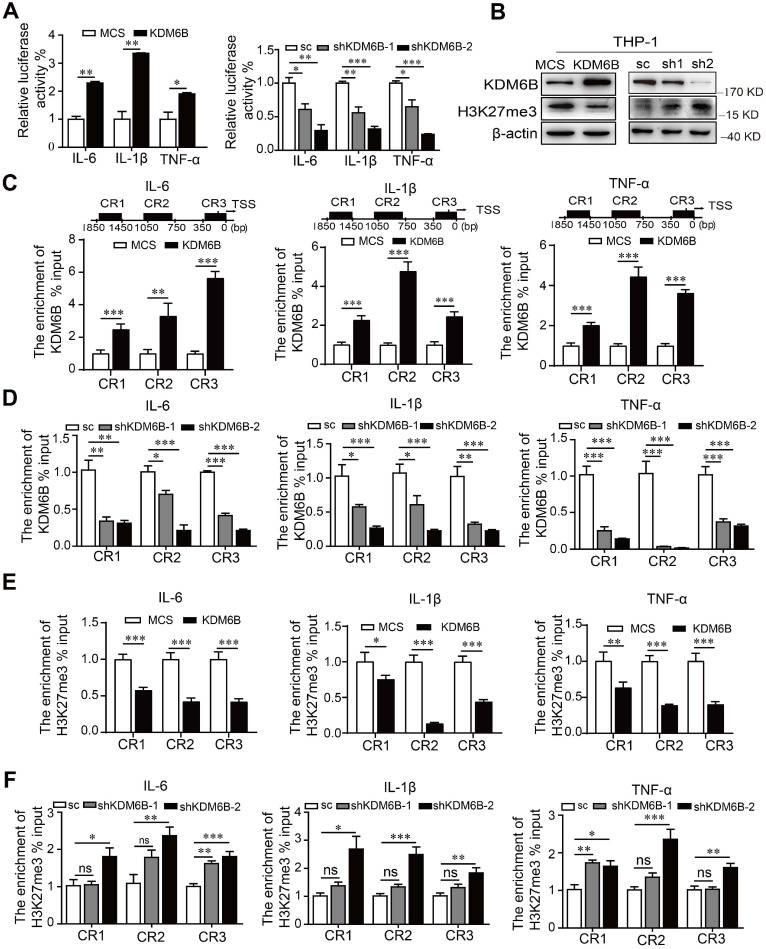
抑制KDM6B的組蛋白去甲基化酶活性會抑制其靶基因的轉錄。因此,使用雙熒光素酶測定來分析M1靶基因的啟動子活性。KDM6B 的過表達特異性刺激了編碼促炎因子 IL-6、IL-1β 和 TNF-α 的基因的啟動子活性。相反,沉默 KDM6B 導致其啟動子活性受到抑制,并且KDM6B在THP-1細胞中的過表達降低了H3K27me3的水平,當KDM6B表達受到抑制時,H3K27me3 的水平會增加。ChIP 分析顯示,與對照相比,THP-1 細胞中KDM6B 的過表達或敲低分別顯著增加或減少了 KDM6B 在這些啟動子的不同區域的占有率。與這些發現一致,當KDM6B在THP-1細胞中過表達或下調時,H3K27me3 向這些啟動子的募集減少或增加。因此,KDM6B 的下調增加了 H3K27me3 的水平和編碼促炎因子的基因的轉錄活性,從而抑制了 M1 極化。
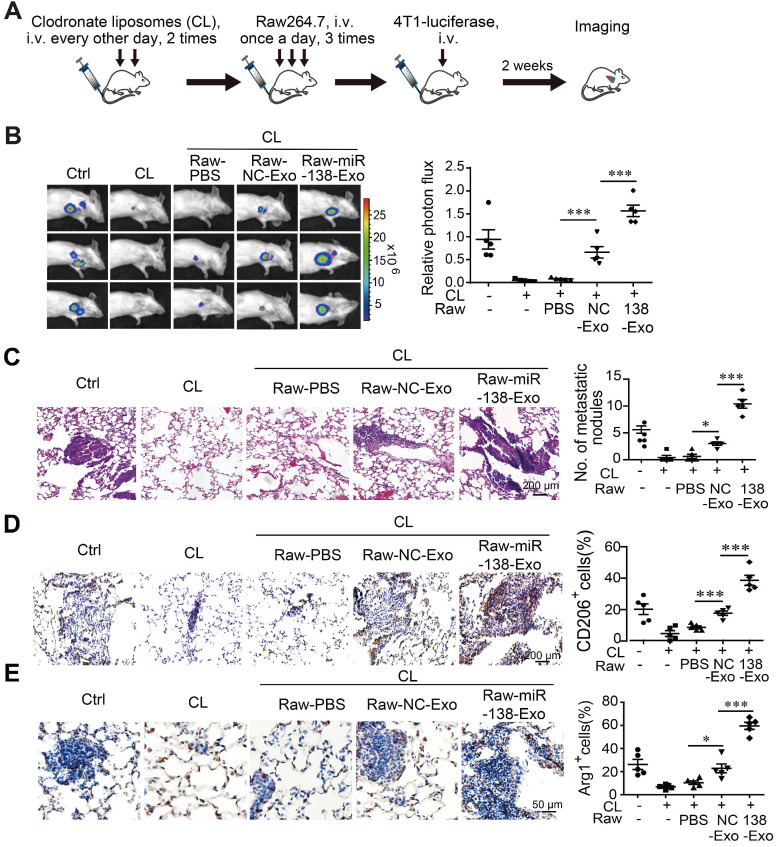
外泌體miR-138-5p促進乳腺癌向肺轉移
極化巨噬細胞在決定腫瘤細胞的表型方面起著重要作用。因此,用外泌體 miR-138-5p處理的M2巨噬細胞是否有助于乳腺癌小鼠異種移植模型中的腫瘤轉移。使用氯膦酸鹽去除小鼠的巨噬細胞,然后轉移用源自T47D或過表達miR-138-5p 的T47D細胞的外泌體處理的Raw264.7細胞。通過尾靜脈靜脈注射4T1-熒光素酶細胞建立小鼠模型。使用體內成像系統設施 (IVIS) 系統監測轉移并計數肺中的轉移性結節。與對照外泌體相比,用T47D-miR-138-5p外泌體處理的巨噬細胞移植小鼠的肺轉移發生率顯著更高。當確定腫瘤組織中CD206和 Arg1陽性M2巨噬細胞的百分比時,發現使用 T47D-miR-138-5p 外泌體處理的巨噬細胞增加了 M2 細胞的百分比。總之,這些結果表明外泌體miR-138-5p誘導的M2巨噬細胞促進了乳腺癌的肺轉移。