






腫瘤抑制因子DRD2-乳腺癌治療靶點
乳腺癌(BrCa)是世界范圍內最常見的癌癥,診斷為IV期患者的5年相對生存率有所下降。晚期BrCa被認為是不可治愈的,目前仍缺乏有效的治療策略。識別和表征新的腫瘤抑制基因對于建立有效的晚期BrCa預后生物標志物或治療靶點非常重要。2021年3月發表于Theranostics(IF=11.556)的文獻“Tumor suppressor DRD2 facilitates M1 macrophages and restricts NF-κB signaling to trigger pyroptosis in breast cancer”對此展開了研究。在本文中,在BrCa中,DRD2被發現由于啟動子甲基化而下調。DRD2的高表達與更長的生存時間正相關,尤其是在HER2陽性患者中。DRD2還能促進BrCa細胞對紫杉醇的敏感性。DRD2異位表達顯著抑制BrCa腫瘤發生。在體內和體外,DRD2也能誘導細胞凋亡和壞死。DRD2通過與β-arrestin2、DDX5和eEF1A2相互作用抑制NF-κB信號通路的激活。有趣的是,DRD2還調節微環境,促進巨噬細胞M1極化,并觸發GSDME執行的焦亡。

技術路線

結果
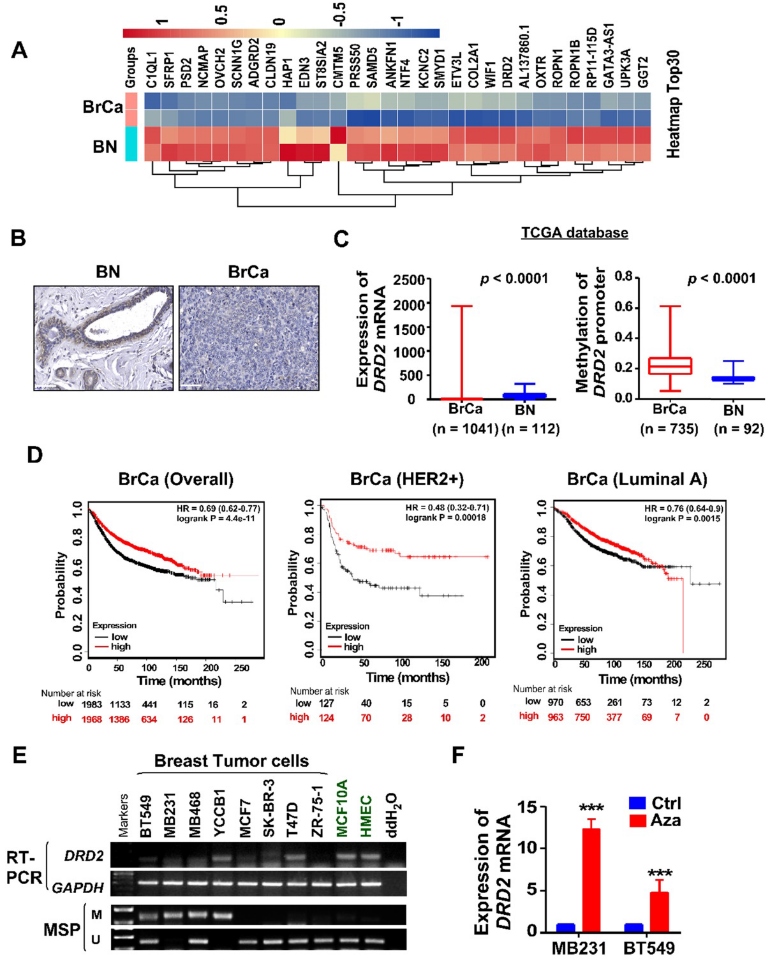
1)DRD2通過啟動子甲基化在轉錄上下調
為了研究新的潛在TSGs,采用BrCa組織和正常組織進行RNA-seq篩選。與正常乳腺組織相比,BrCa組織中DRD2 mRNA表達明顯下調(圖1A)。免疫組化染色發現,與BrCa組織相比,正常乳腺組織中DRD2蛋白水平也較高(圖1B)。同樣,基于TCGA,在BrCa中也觀察到DRD2 mRNA的下調,并且在BrCa中DRD2啟動子甲基化也更頻繁(圖1C)。根據Kmplot,DRD2的高表達促進了BrCa患者的更長的生存期,這也在HER2陽性基因型患者中可見。但這一優勢在Luminal A患者中未見(圖1D)。根據RT-PCR和MSP結果,幾乎所有的BrCa細胞系與永凍的正常乳腺細胞系相比,都可以看到DRD2 mRNA表達下調或缺失以及啟動子甲基化(圖1E)。因此,DRD2的高甲基化頻率可能有助于其下調BrCa。Aza藥物去甲基化恢復了兩種沉默的BrCa細胞系MDA-MB231和BT549中DRD2的表達(圖1F)。
2)在體內和體外,DRD2的表達抑制BrCa細胞的腫瘤發生
構建過表達DRD2的MDA-MB231和BT549細胞,通過RT-PCR (Figure 2A)和WB (Figure 2B)驗證。CCK8實驗證明,DRD2異位表達抑制腫瘤細胞生長(圖2C)。在單層集落形成實驗(圖2D)和軟瓊脂形成實驗(圖2E)中,DRD2也損害了存活能力。并進行細胞凋亡和細胞周期分布分析。DRD2的表達顯著促進了BrCa細胞凋亡(圖2F),并阻斷了MDA-MB231和BT549在G2/M期的凋亡(圖2G)。此外,過表達DRD2促進了BrCa細胞對PTX的敏感性(圖2H)。在傷口愈合實驗中,異位表達的DRD2比對照組表現出更少的閉合人工傷口的能力(圖2I)。與此相一致的是,DRD2的過表達顯著降低了BrCa的遷移和侵襲(圖2J)。在體內進一步測定了DRD2的抑瘤作用。皮下腫瘤模型顯示過表達DRD2模型的腫瘤體積和重量均減小(圖2K)。

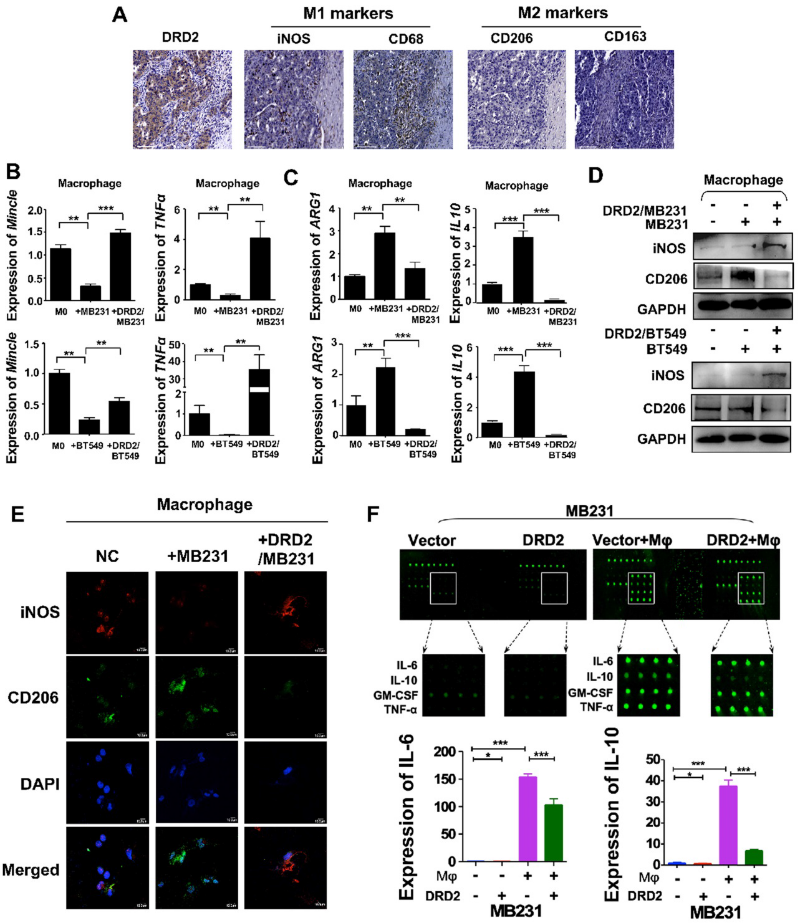
3)DRD2重新編碼Mφ M1表型,下調IL-6和IL-10
據報道,DRD2可調節M1的極化。結果顯示,在DRD2表達水平較高的BrCa組織中,M1 Mφ的浸潤增加,M2 Mφ的浸潤減少(圖3A)。為進一步研究DRD2對TAMs的調控作用,構建了BrCa與Mφ共培養體系。當Mφ與表達DRD2的BrCa細胞共培養時,qRT-PCR顯示M1表型標記增加,M2 Mφ標記下調(圖3B-C)。qRT-PCR也證實了載體轉染的BrCa細胞將Mφ調節為M2型(圖3C)[12]。WB結果顯示,表達DRD2的BrCa細胞上調M1標記iNOS,下調M2標記CD206(圖3D)。IF染色顯示,與表達DRD2的腫瘤細胞共培養后,Mφ表現為M1表型(圖3E)。上述結果表明,BrCa中的DRD2具有將Mφ重新編碼為M1表型的能力。
為了探索使Mφ向M1表型分化的關鍵調控因子,我們進行了細胞因子陣列分析。結果表明,TNF-α和兩種誘導M1極化的經典細胞因子IFN γ均未增加。而與Mφ共培養后,DRD2顯著下調IL-6和IL-10(圖3F)。分析熒光值,進一步證實IL-6和IL-10下調(圖3F)。因此,DRD2可以將Mφ重新編碼為M1表型,并在crosstalk過程中顯著下調IL-6和IL-10。
4)DRD2重編程的Mφ誘導腫瘤細胞的焦亡
如HE所示,來自小鼠模型的腫瘤承載DRD2顯示腫瘤細胞死亡增加(圖4A)。在免疫組化染色中,表達DRD2的4T1腫瘤樣本中pMLKL表達較高(圖4A)。根據TUNEL實驗,DRD2也促進了體內細胞凋亡(圖4B)。DRD2上調GSDME而不是GSDMD(圖4C)。在crosstalk過程中,Mφ進一步促進了DRD2轉染的BrCa細胞中GSDME的表達(圖4C)。在表達DRD2的BrCa細胞中,Mφ也能上調IL-1β(圖4C)。WB結果表明,DRD2誘導了MLKL的磷酸化,而在共培養過程中,MLKL被Mφ抑制(圖4D)。此外,DRD2表達激活了caspase-8,而激活被Mφ抑制(圖4D)。假設,DRD2可能在與Mφ crosstalk時誘發焦亡。BrCa細胞中與Mφ共培養時,NLRP3在表達DRD2的BrCa細胞中被觸發。激活的caspase-1蛋白水解也能誘導IL-1β和IL-18的成熟(圖4D)。在共培養過程中,Cleaved caspase-3表達上調(圖4D)。此外,在共培養過程中發現表達DRD2的BrCa細胞中,GSDME的N端發生了剪切(圖4D)。為了確定是否由M1 Mφ引起焦亡,我們使用了LPS誘導的M1 Mφ培養基,結果表明,在表達DRD2的BrCa細胞中,M1 Mφ僅觸發NLRP3組裝并剪切GSDME(圖4E)。以上結果提示, M1 Mφ以DRD2依賴的方式觸發BrCa細胞的焦亡。

5)DRD2通過阻斷TAK1的磷酸化來限制NF-κB信號的激活
NF-κB信號的激活對于觸發炎癥小體組裝和隨后的焦亡是必不可少的。IF染色顯示DRD2幾乎阻斷了p-p65的核易位(圖5A),但這種抑制被Mφ所抵消(圖5B)。核和細胞質提取物也顯示DRD2抑制了p-p65的核易位(圖5C)。如WB結果所示,在LPS刺激下,DRD2抑制IKKα/β、IκBα和p65的磷酸化(圖5D)。異位DRD2表達也顯著抑制了Mφ誘導的p65磷酸化和IKKα/β的上游激活(圖5E)。WB結果顯示DRD2下調NF-κB的下游靶點ICAM-1。然而,這種下調被Mφ所抵消(圖5E)。抑制IKKα/β磷酸化提示DRD2負向介導IKK復合物上游。TAK1在催化IKKα和IKKβ中起關鍵作用。TAK1的磷酸化被異位DRD2表達顯著抑制(圖5D-E)。因此,DRD2通過阻斷上游激酶TAK1來抑制NF-κB信號通路的激活。
作為一種典型的G蛋白偶聯受體, DRD2的內化誘導其受體β-arrestin2在質膜上的募集,并增加其與β-arrestin2的親和力。LPS和CM刺激了DRD2和β-arrestin2的蛋白-蛋白結合(圖5F-G)。同時,通過IF染色觀察到,經過LPS處理后,DRD2似乎在細胞質中與p-p65結合(圖5A), Co-IP和IB進一步證實了這種結合(圖5F)。據報道,β-arrestin2信號的激活會破壞星形膠質細胞中TAK1-Tab1的結合,而這種結合對于TAK1的激活至關重要。本研究還證實,內化的DRD2可以促進TAB1與β-arrestin2的結合,削弱TAB1與TAK1的結合(圖5H)。綜上所述,DRD2激活的β-arrestin2通過競爭性地結合TAB1來拮抗TAK1的磷酸化。

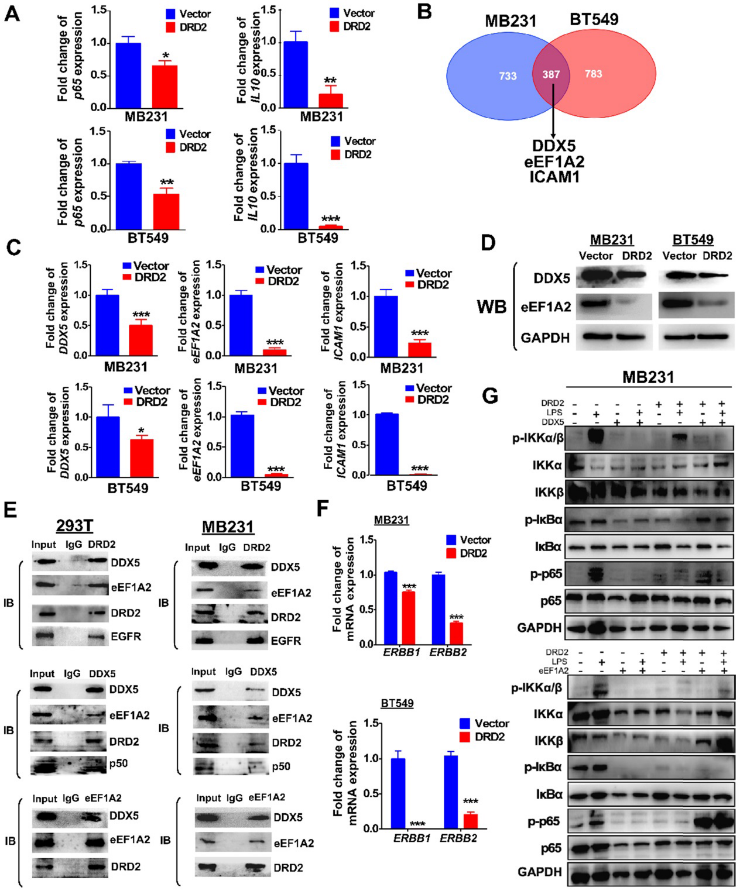
6)DRD2通過下調DDX5和eEF1A2來抑制NF-κB通路的激活和腫瘤的發生
DRD2異位表達顯著抑制了p65和NF-κB靶基因IL-10的mRNA表達(圖6A),表明在沒有配體激活的情況下,DRD2是NF-κB通路的負調控因子。除了結合激活β-arrestin2外,DRD2還可能通過其他方式抑制NF-κB信號通路。采用Co-IP法分離可能的結合蛋白,并采用質譜法進行蛋白鑒定。MDA-MB231和BT549細胞的所有結合蛋白中,DDX5、eEF1A2和ICAM-1均被異位表達的DRD2下調(圖6B-C)。根據以往的研究,DDX5和eEF1A2是幾種癌癥中的兩種致癌基因。WB也證實了DDX5和eEF1A2蛋白水平下調(圖6D)。在293T和MDA-MB231中,通過Co-IP和IB實驗證實了DRD2、DDX5和eEF1A2的結合(圖6E),表明這三種蛋白形成了一個復合物。根據以往的研究,DDX5可以結合p50,并協助IκB釋放p50。本研究還揭示了293T和MDA-MB231中DDX5與p50的結合(圖6E)。此外,有報道稱DRD2可在神經系統中與EGFR結合。在BrCa細胞中,293T和MDA-MB231中也證實了DRD2和EGFR的結合(圖6E)。DRD2的表達下調ERBB1 (EGFR)和ERBB2 (HER2)的表達(圖6F)。在異位表達DRD2的BrCa細胞中,DDX5可以促進p-IκBα的磷酸化,增加磷酸化p65的蛋白水平(圖6G)。eEF1A2可直接顯著上調p-p65的表達,而不影響IKKα/β或IκBα,甚至在沒有LPS的情況下,eEF1A2可上調表達DRD2的MDA-MB231的p-p65蛋白水平(圖6G)。以上結果表明,DDX5和eEF1A2對NF-κB信號通路的促進作用受DRD2異位表達的抑制。
結論:這項研究新發現了一種腫瘤抑制基因-DRD2,可以提高BrCa患者的生存率和PTX治療反應。DRD2誘導細胞凋亡和壞死,并在Mφ向M1重編程過程中進一步觸發焦亡。DRD2通過與β-arrestin2結合,下調DDX5和eEF1A2,從而限制NF-κB信號通路的激活。DRD2是一種潛在的預測預后的生物標志物,并且DRD2是BrCa中一個很有前途的治療靶點。