






國自然熱點之焦亡
細胞焦亡(Pyroptosis)是一種炎癥細胞程序性死亡方式,主要通過炎癥小體介導包含半胱天冬酶-1(Caspase-1)在內的多種Caspase的激活,造成包括GSDMD在內的多種Gasdermin家族成員發生剪切和多聚化,造成細胞穿孔,進而引起細胞死亡。近些年細胞焦亡的中標數一直在上升,特別是2018和2019,這兩年是呈現直線上升。接下來,我們通過一篇文獻來了解焦亡研究的套路。
這篇題為“Neural progenitor cell pyroptosis contributes to Zika virus-induced brain atrophy and represents a therapeutic target”的文章講述了ZIKV破壞神經發生并導致小頭畸形的機制。在本研究中,我們發現ZIKV通過誘導caspase-1和 GSDMD介導的焦亡直接影響神經祖細胞的發育,將ZIKV感染與小頭畸形的發生聯系起來。重要的是,caspase- 1缺失或其抑制劑VX-765治療可減少ZIKV誘導的炎癥反應和焦亡,顯著減輕體內神經病理學和腦萎縮。
結果:
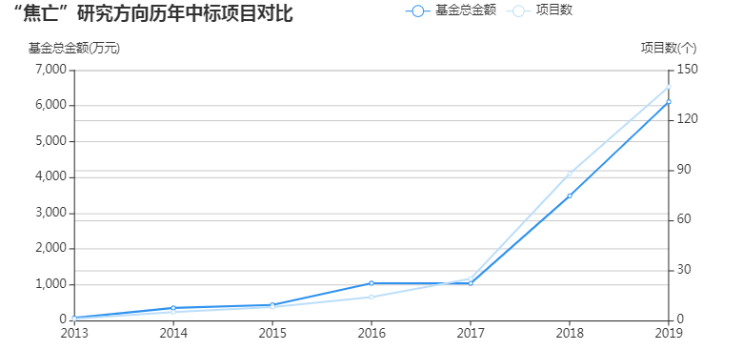
1) ZIKV在新生鼠腦中復制并導致嚴重的神經病理學改變和腦萎縮
為了評估ZIKV感染在小鼠腦發育過程中是否具有致病性,WT幼鼠皮下感染5×105個ZIKV PFU。值得注意的是,在從感染后3dpi到21 dpi期間,ZIKV感染的小鼠顯示出比模擬感染的小鼠更低的體重(圖1A)。此外,在ZIKV感染的小鼠中有明顯的腦萎縮,表現為在21 dpi時背側腦面積減少(圖1 B和1C)。使用qRT-PCR,證實在ZIKV感染小鼠的大腦中存在大量病毒核糖核酸拷貝,表明觀察到的腦萎縮與ZIKV感染密切相關(圖1D)。為了進一步研究ZIKV感染對腦發育的可能致病作用,對小鼠腦的組織切片進行了神經病理學檢查。值得注意的是,ZIKV感染的小鼠大腦顯示出大量中性粒細胞浸潤、壞死和主要在海馬、丘腦和皮層區域的正常細胞結構的破壞,表明大腦中存在嚴重的局灶性炎癥,而模擬感染不會引起病理變化(圖1 E和F)。此外,ZIKV感染小鼠的大腦經常出現單核細胞和小膠質細胞的血管周圍套袖。綜上所述,我們的發現表明ZIKV感染導致腦萎縮,并在所用的小鼠感染模型中誘導強烈的炎性腦損傷。
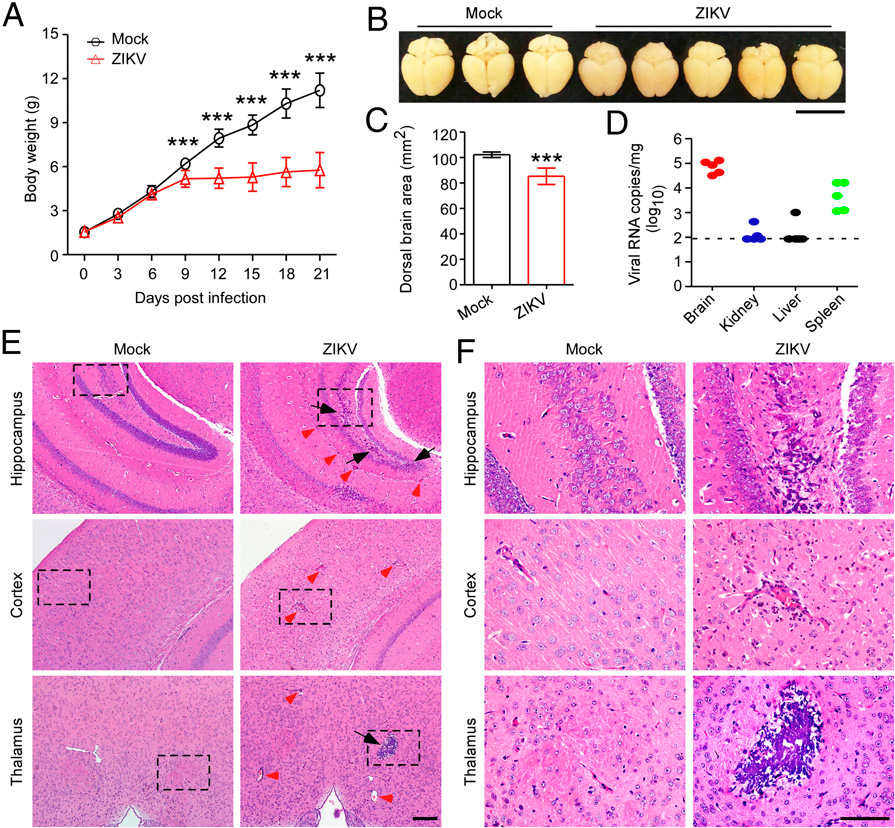
2) ZIKV感染導致體內NPC焦亡
基于我們前述對寨卡病毒感染小鼠大腦炎癥損傷的觀察,我們進一步探索ZIKV感染是否也能在NPC人群的大腦中引發caspase-1介導的焦亡。如圖2 A和圖2B所示,當在具有指示標記的NPC小生境中對裂解的caspase-1 (p10)和PI進行染色時,感染寨卡病毒的小鼠顯示出豐富的裂解的caspase-1和PI+細胞。
由于焦亡與炎癥小體激活有聯系,接下來在從ZIKV感染和模擬感染小鼠獲得的腦組織標本中評估了更廣泛的炎癥小體基因。如圖2C所示,在小鼠腦組織標本中可檢測到所有與炎癥體相關的基因,與模擬感染相比,寨卡病毒感染的小鼠中IL1B、IL-18、CASP1、ASC和GSDMD轉錄物的水平顯著升高。此外,與模擬對照相比,NLRP3在ZIKV感染的腦中的表達顯著增加 (圖2D)。在ZIKV感染的大腦中,caspase-1和GSDMD的裂解程度都很高(圖2E)。綜上所述,這些數據支持了ZIKV感染NPCs并在體內引發焦亡的觀點。
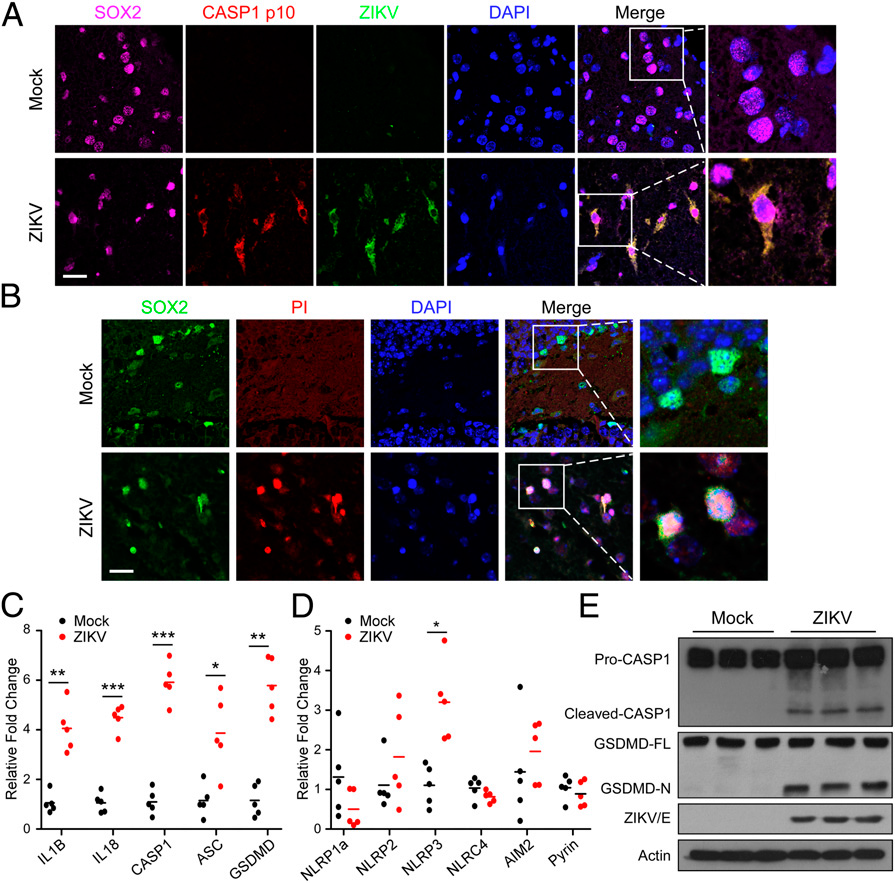
3) ZIKV在體外感染人NPCs并誘導焦亡
為了進一步了解ZIKV和神經發生障礙之間的可能聯系,我們接下來評估了ZIKV誘導的人NPCs(hNPCs)的焦亡的生物學效應。在ZIKV感染的hNPCs中,出現典型的炎性改變,包括細胞腫脹和膜破裂(圖3A)。hNPCs中的ZIKV感染通過細胞中ZIKV E抗原和SOX2的共同存在被證實(圖3B)。此外,通過透射電子顯微鏡(TEM)進行的超微結構分析也揭示了壞死的形態學變化,顯示了ZIKV感染后染色質聚集、線粒體腫脹和質膜破裂(圖3C)。我們接下來檢查了細胞裂解物中caspase-1的活性,數據顯示受ZIKV感染的細胞中caspase-1活性增加(圖3D)。此外,用PI/Hoechst 33342雙染色法進一步驗證了炎癥壞死細胞,正如所料,感染ZIKV的hNPCs發生了焦亡(圖3 E和F)。此外,與模擬感染相比,ZIKV感染顯著增加了乳酸脫氫酶的釋放量,而與siRNA陰性對照組相比,沉默caspase-1的表達減少了ZIKV感染引起的乳酸脫氫酶的釋放量(圖3G),這表明caspase-1介導的NPCs的焦亡有助于ZIKV發病機制。與模擬感染的對應細胞相比,ZIKV感染觸發了hNPCs中caspase-1和GSDMD的裂解(圖3H)。 
4) ZIKV誘導人類神經球的焦亡
為了進一步說明神經發生過程中ZIKV感染的生物學效應,我們接下來用ZIKV感染神經細胞的3D培養物,然后觀察神經球的形成。我們發現模擬感染的hNPCs產生圓形神經球,而ZIKV感染的神經球表現出形態學異常,并有焦亡的跡象(圖4 A和4B)。此外,模擬感染的神經球隨著時間的推移繼續生長,但只有少數ZIKV感染的神經球以較小的尺寸存活(圖4A-D)。為了進一步證實感染ZIKV的神經球發生了焦亡,我們用活性caspase-1和PI對神經球進行了雙重染色。如圖4E所示,感染ZIKV的神經球含有染色強烈的活性caspase-1和PI,這表明ZIKV感染可以誘導神經球的焦亡。神經球感染ZIKV通過神經球中存在ZIKV E抗原和SOX2被證實(圖4F),此外,還測定了ZIKV感染后神經球的乳酸脫氫酶釋放,表明ZIKV感染誘導了神經球的乳酸脫氫酶釋放(圖4G)。

5) Caspase-1的缺失逆轉ZIKV誘導的神經病理學改變和腦萎縮
我們生成了caspase-1-null小鼠,以進一步評估焦亡ZIKV誘導的腦畸形發病機制中的作用。我們發現與模擬感染組相比,感染ZIKV的WT小鼠體重較低,而ZIKV感染的Casp1?/?小鼠體重與模擬感染組在21日齡時的體重相似(圖5A)。與ZIKV感染的野生型小鼠相比,ZIKV感染的Casp1?/?小鼠的嚴重腦萎縮被徹底消除(圖5 B和5C)。組織病理學檢查進一步表明,與ZIKV感染的野生型小鼠相比,Casp1?/?小鼠都免于炎癥誘導的損傷(圖5D)。此外,當在腦組織中測量促炎細胞因子IL-1β和IL-18的表達水平時,我們發現與野生型小鼠相比,在感染ZIKV的小鼠中,caspase-1的減少消除了IL-1β和IL-18的增加(圖5 E和F)。此外,我們的結果還表明,ZIKV感染引發了GSDMD的裂解,而caspase-1缺失減弱了GSDMD的裂解(圖5G)。因此,由ZIKV引起的神經病理學和腦萎縮可以通過廢除caspase-1介導的焦亡來抑制。

6) VX-765治療減輕ZIKV感染小鼠的神經病理學和腦萎縮
caspase-1在介導ZIKV相關神經病理學中的重要作用促使我們研究caspase-1是否可以成為神經病理學和小頭畸形的潛在治療靶點。VX-765是是一種強效和選擇性的caspase-1抑制劑。結果表明,對照載體或Z-DEVD-FMK處理的小鼠比模擬感染的對照小鼠增重慢,而VX-765處理的小鼠沒有出現這種結果(圖6A)。30天后,ZIKV感染小鼠的存活率僅為38%,相比之下,VX-765治療的小鼠的存活率更高,為69%。盡管用Z-DEDFMK治療的所有小鼠在感染后第16天死亡(圖6B),但進一步的觀察表明,Z-DEVD-FMK治療的小鼠與用載體或VX-765治療的小鼠在沒有ZIKV感染的情況下在體重變化和存活率方面沒有顯著差異。此外,與載體對照處理的ZIKV感染小鼠相比,VX-765處理減輕了由ZIKV感染引起的嚴重腦萎縮(圖6 C和6D)。
進一步的組織病理學檢查顯示,在ZIKV感染的對照組和Z-DEVD-FMK治療的小鼠的海馬中觀察到大量的中性粒細胞浸潤和壞死位點,但在VX-765治療后顯著減弱(圖6E)。免疫組織化學(IHC)對小膠質細胞和星形膠質細胞的Iba-1和GFAP特異性標記物的分析分別用于評估大腦中的神經炎癥。如圖6F所示,與模擬感染小鼠相比,ZIKV感染對照組和Z-DEVD-FMK治療組小鼠的海馬體中的Iba-1和GFAP水平顯著更高。值得注意的是,VX-765的治療降低了Iba-1和GFAP的表達水平,與未感染小鼠的大腦相當,這表明VX-765能夠逆轉神經炎癥。
此外,在暴露于ZIKV的對照載體處理的小鼠的腦組織中,促炎細胞因子IL-1β和IL-18的表達水平上調,而這種上調被VX-765處理抑制,但不被Z-DEVD-FMK抑制(圖6 G和H)。一致地,在VX-765治療后,ZIKV感染小鼠的大腦中caspase-1和GSDMD的蛋白酶裂解受到抑制,這表明VX-765抑制了體內與ZIKV感染相關的焦亡(圖6I)。
