國自然熱點-自噬
2020年國自然已塵埃落定,2021年國自然申請又即將開始。那么接下來小編給大家介紹一下國自然熱點之一—自噬。自噬從2010到2017申請的相關項目一直在上升,近兩年稍有回落,但也多達500多項。其中腫瘤學相關的研究遙遙領先。話不多說,今天和大家分享一篇自噬相關的文章。


這篇發表于影響因子10.717的“Cell Death & Differentiation”雜志的文章“DAPK3 inhibits gastric cancer progression via activation of ULK1-dependent autophagy” 報道了DAPK3在胃癌細胞中誘導自噬的抑癌作用。DAPK3在GC中的抑癌作用與自噬相關,DAPK3是一種新型的自噬調節因子,可以直接磷酸化ULK1并激活ULK1。因此,DAPK3可能是一個潛在的預后的自噬相關標志物。
結果:
1)DAPK3在胃癌細胞中顯示腫瘤抑制活性
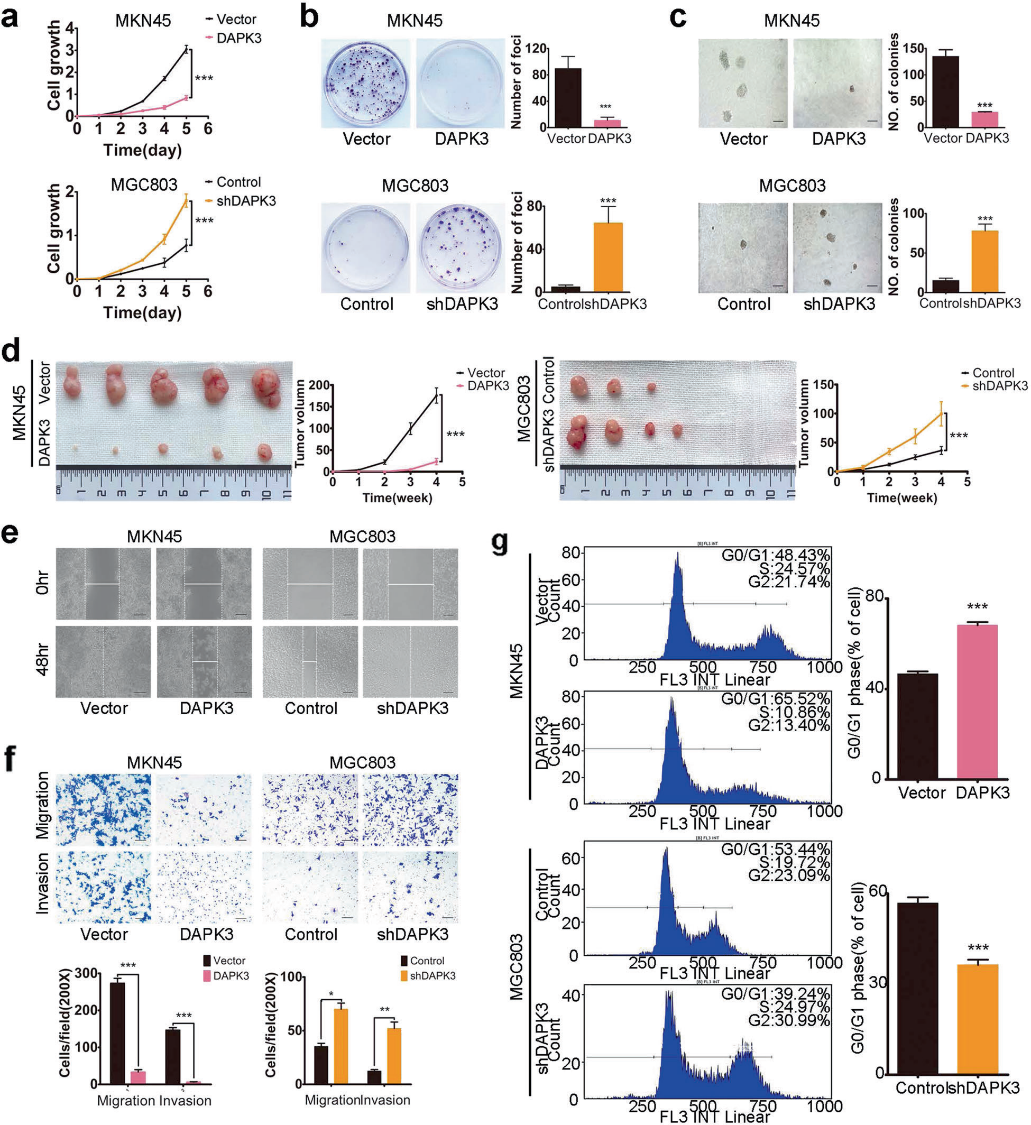
我們前期研究表明DAPK3的表達與胃癌晚期臨床分期和不良預后呈負相關。為了深入了解DAPK3在胃癌細胞中的腫瘤抑制功能,我們進行了以下實驗。 XTT增殖試驗顯示DAPK3顯著抑制腫瘤細胞生長速率(圖1a)。病灶形成和軟瓊脂試驗顯示DAPK3顯著降低了依賴錨定和獨立細胞中的集落頻率和大小(圖1b,c)。MGC803和GES-1細胞中DAPK3的敲除強烈促進細胞生長和集落形成(圖1a-c)。為了驗證DAPK3對體內腫瘤生長的影響,建立了皮下異種移植瘤小鼠模型。我們發現,空載體轉染的MKN45細胞在注射后7天內就可以看到腫瘤,而DAPK3轉染的MKN45細胞直到注射后3周才觀察到腫瘤(圖1d)。動力學圖顯示由DAPK3沉默的MGC803細胞產生的腫瘤明顯大于來自對照細胞的腫瘤(圖1d)。傷口愈合和transwell試驗表明,DAPK3過表達后細胞遷移和侵襲明顯減少,DAPK3敲除后細胞遷移和侵襲增加(圖1e,f)。另外,與對照細胞相比,DAPK3過表達的MKN45和MKN28細胞顯示出G0/G1期細胞百分比的顯著增加。同時,當DAPK3沉默時,我們觀察到MGC803和GES-1細胞中G0/G1期細胞的百分比顯著降低(圖1g)。
2)氨基酸剝奪誘導自噬需要DAPK3
我們研究了DAPK3是否調節胃癌細胞的自噬。蛋白質印跡分析顯示,EBSS處理促進自噬后,內源性DAPK3蛋白水平顯著增加(圖2a)。類似地,DAPK3過表達增加了LC3-II/LC3-I比率。同時,在氨基酸饑餓條件下,SQSTM1/p62水平在DAPK3過表達的細胞中顯著降低 (圖2b)。此外,在DAPK3過表達的細胞中發現LC3的數量增加,表明DAPK3增強了自噬體的形成(圖2c)。透射電鏡顯示,與對照細胞相比,DAPK3過表達細胞中含有細胞質結構和殘留消化物質的自噬空泡顯著增加(圖2d)。MGC803和GES-1細胞中DAPK3的敲除顯著損害了LC3-I向LC3-II的轉化、SQSTM1/p62的降解、GFP-LC3的積累和氨基酸饑餓期間自噬空泡的形成(圖2d)。因此,DAPK3是誘導氨基酸饑餓介導的自噬所必需的。

3) ATG5或ATG7缺失抑制的自噬會削弱DAPK3的腫瘤抑制功能
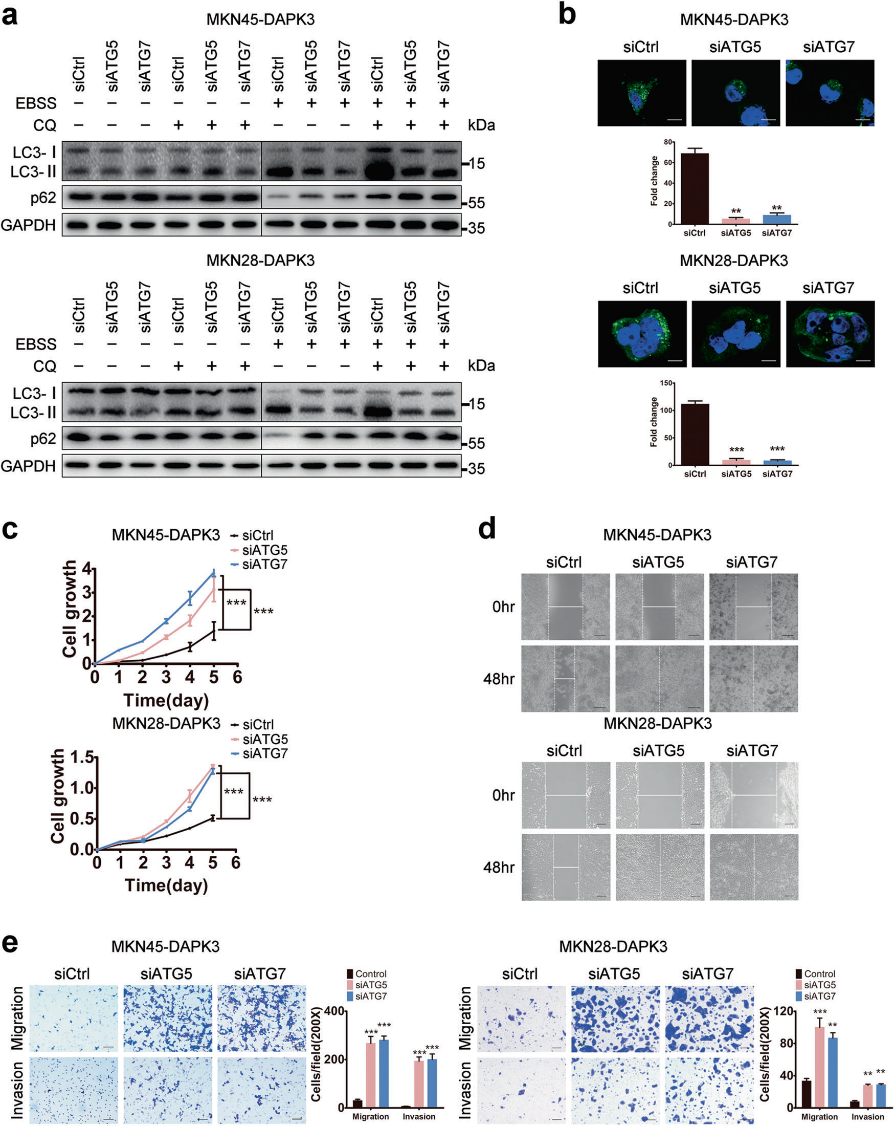
為了確定DAPK3介導的自噬是否與其腫瘤抑制功能相關,在DAPK3過表達細胞和空載體轉染細胞中,抑制ATG5或ATG7。ATG5或ATG7的敲除顯著降低了DAPK3過表達細胞中LC3-I向LC3-II的轉化、SQSTM1/P62降解和GFP-LC3形成,表明DAPK3誘導的自噬受到ATG5或ATG7耗竭的損害(圖3a,b)。接下來,我們研究了自噬抑制對DAPK3過表達細胞和空載體轉染細胞的細胞增殖、遷移和侵襲的影響。細胞生長試驗顯示細胞生長速率在ATG5或ATG7缺失的DAPK3過表達細胞明顯高于對照組細胞。同時,ATG5或ATG7敲除促進細胞遷移和侵襲(圖3c-e)。這些結果表明,ATG5或ATG7敲除可以挽救DAPK3誘導的GC細胞的生長和轉移阻滯。
4) DAPK3通過ULK1激活調節自噬
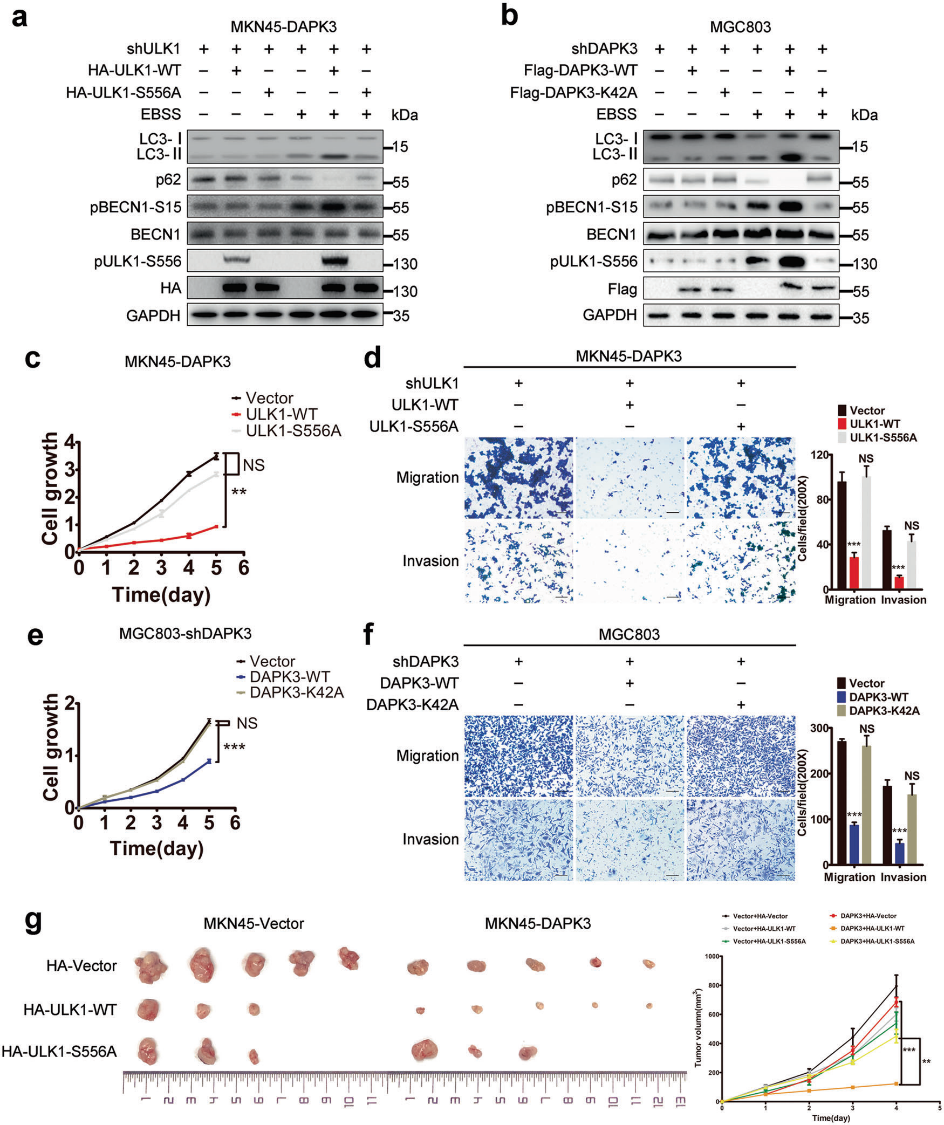
為了闡明DAPK3介導的自噬的分子機制,我們使用基于質譜的定量磷酸化蛋白質組分析來分析在氨基酸饑餓下DAPK3過表達的MKN28細胞和對照細胞中差異表達的蛋白質。與對照細胞相比,我們觀察到DAPK3過表達細胞中Ser556的ULK1磷酸化增加了大約兩倍。免疫印跡分析顯示氨基酸饑餓后DAPK3過表達細胞中Ser556上ULK1磷酸化水平的顯著上調(圖4a)。我們還檢測了Beclin-1的磷酸化狀態,Beklin-1是ULK1的底物,是VPS34復合物激活和完全自噬誘導所必需的。正如預期的那樣,DAPK3增加了Ser15的Beclin-1磷酸化,表明DAPK3調節ULK1激活(圖4a)。此外,ULK1敲除強烈抑制自噬通量、自噬體形成,并增強DAPK3過表達細胞在體外的致瘤性(圖4b-g)。在接種了ULK1沉默的MKN45-DAPK3細胞的裸鼠中,腫瘤大小顯著增加,表明DAPK3介導的腫瘤抑制依賴于ULK1的激活(圖4h)。

5) DAPK3通過Ser556的直接磷酸化激活ULK1
為了研究DAPK3直接磷酸化ULK1的可能性,我們使用了GPS軟件來預測DAPK3的ULK1磷酸化位點,并發現ULK1可能在Ser556被DAPK3磷酸化(圖5a)。DAPK3在Ser556磷酸化重組ULK1,提示DAPK3可以直接在Ser556磷酸化ULK1(圖5b)。DAPK3還在體外激酶測定中強烈磷酸化Ser556 位點的ULK1,在氨基酸饑餓狀態下,ULK1的Ser556位點磷酸化水平進一步提高(圖5c)。然而,Ser556突變顯著阻礙了該位點的磷酸化(圖5c)。為了進一步驗證體內ULK1中DAPK3磷酸化位點,我們通過免疫沉淀來檢測Ser556位點ULK1磷酸化。正如預期的那樣,在DAPK3過表達的細胞中觀察到高水平的ULK1 Ser556磷酸化(圖5d)。WT DAPK3過表達導致的Ser556 ULK 1磷酸化的增加明顯高于DAPK3 K42A過表達導致的增加,表明DAPK3激酶活性是ULK1 Ser556磷酸化所必需的(圖5e)。
接下來,我們將HA-ULK1作為關鍵分子進行免疫沉淀,并分析了在DAPK3過表達細胞和空載體對照細胞中ULK1和其他ULK1復合物成分之間的相互作用。DAPK3表達在饑餓條件下顯著增強了WT ULK1與其結合配偶體FIP200、ATG13和ATG101之間的相互作用,而ULK1中Ser556磷酸化位點的缺失部分削弱了這些相互作用(圖5d)。ATG101在anti-Flag免疫沉淀中被發現,饑餓后WT DAPK3細胞中ATG101的量增加。饑餓時,FIP200和ATG13與標記的WT DAPK3共免疫沉淀(圖5e)。ULK1通過直接磷酸化Beclin1和ATG14L,有利于VPS34復合物的激活。于是我們通過蛋白質印跡分析探討了DAPK3在VPS34復合物中的作用。研究表明,在饑餓條件下激活HEK293T細胞中的VPS34復合物需要ULK1 Ser556的磷酸化和DAPK3激酶活性(圖5f,g)。總的來說,這些數據表明DAPK3對ULK1的磷酸化增強了ULK1復合物的形成和饑餓時VPS34復合物的活化。

6) ULK1中的Ser556磷酸化促進DAPK3介導的自噬和腫瘤抑制
為了確定ULK1磷酸化在DAPK3調節的自噬和腫瘤抑制中的功能意義,我們將WT ULK1或S556A ULK1 cDNA轉染到DAPK3過表達和空載體的MKN45細胞中。DAPK3過表達的MKN45細胞在shRNA誘導的內源性ULK1下調下表現出自噬缺陷和致瘤性增加。WT ULK1 cDNA的重建恢復了自噬和腫瘤抑制(圖6a,c,d)。為了進一步評估UKL1磷酸化對體內腫瘤生長的影響,我們進行了腫瘤異種移植實驗。帶有WT ULK1的DAPK3過表達MKN45細胞的異種移植物生長速度比來自S556A突變表達細胞的異種移植物生長速度慢。與空載體對照細胞相比,WT ULK1表達在DAPK 3過表達的MKN45細胞中顯示出更顯著的腫瘤生長抑制作用(圖6g)。然后我們分析了DAPK3激酶活性對自噬誘導和腫瘤抑制的重要性。與DAPK3 K42A細胞相比,含有DAPK3 WT的MGC803細胞顯示出更高的自噬水平和更低的細胞生長率、運動性和侵襲性(圖6b,e,f)。綜上所述, DAPK3的激酶活性和Ser556的ULK1磷酸化是DAPK3功能所必需的。
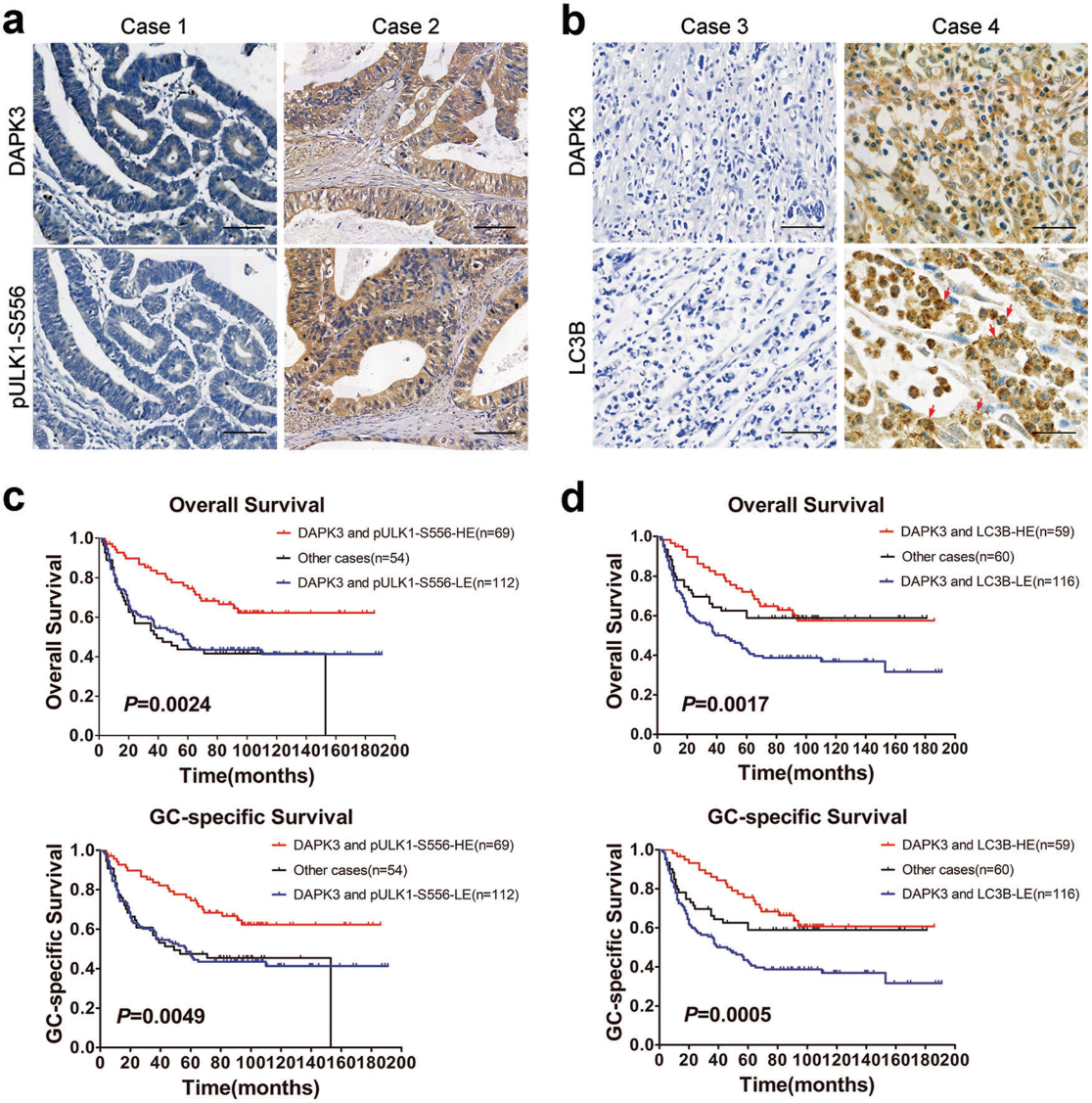
7) DAPK3的表達與手術GC標本中Ser556和LC3B的ULK1磷酸化呈正相關
我們的體外研究表明,DAPK3對ULK1 Ser556的磷酸化對自噬誘導和腫瘤抑制至關重要。相關分析表明DAPK3表達與GC組織中pULK1 Ser556和LC3B的免疫染色呈正相關(圖7a,b)。Kaplan-Meier分析顯示,DAPK3和pULK1 Ser556共表達的患者具有更長的總生存期和癌癥特異性生存期。同樣,DAPK3和LC3B的共同表達預示著患者的良好預后(圖7c,d)。這些數據表明DAPK3相關的自噬對人胃癌的進展有明顯的抑制作用。