






泛素化酶VS巨噬細胞極化
導語:巨噬細胞是先天性免疫系統的效應細胞,對宿主防御、炎癥消退和傷口愈合至關重要。巨噬細胞有促炎性和抗炎性的兩個極端。了解調控巨噬細胞極化的機制,對于宿主防御和修復組織至關重要。泛素特異性蛋白酶19(USP19)是一種內質網(ER)錨定的去泛素化酶,調節ER相關蛋白降解和DNA損傷修復。泛素化酶與巨噬細胞極化的研究值得關注。

(IF=8.484)
技術路線

結果
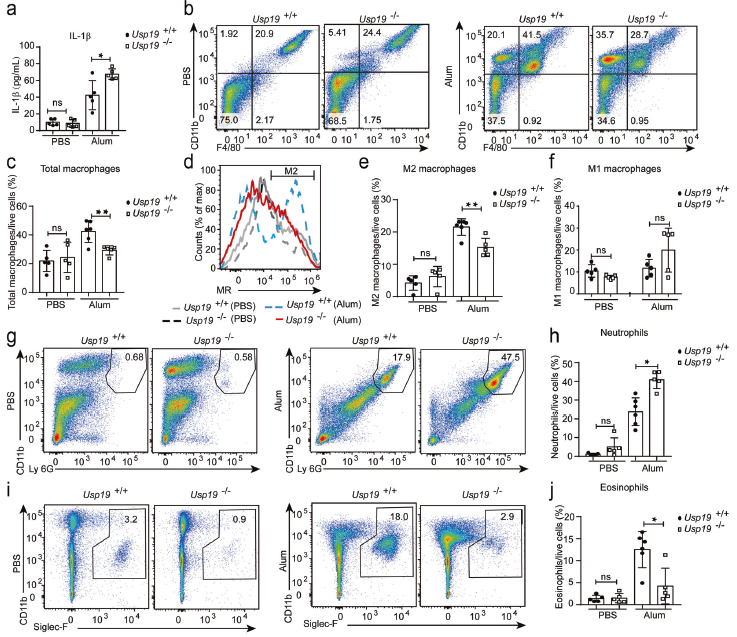
為揭示USP19在炎癥中的作用,使用鋁鹽(明礬)誘導的腹膜炎模型來檢測IL-1β的產生,發現Usp19-/-小鼠的灌洗液中IL-1β的濃度顯著高于野生型小鼠。為確定Usp19-/-小鼠IL-1β產生的增加是否與巨噬細胞募集的增加有關,定量灌洗液中的巨噬細胞。發現明礬處理后Usp19-/-小鼠中巨噬細胞總數減少。因此,推斷Usp19-/-小鼠M1樣巨噬細胞和M2樣巨噬細胞之間的平衡可能發生改變。明礬處理后,Usp19-/-小鼠中M2樣巨噬細胞(F4/80 + CD11b + MR +)而非M1樣巨噬細胞(F4/80 + CD11b + CD86 +)的比例顯著降低。巨噬細胞極化與中性粒細胞和嗜酸性粒細胞募集密切相關,發現Usp19-/-小鼠中明礬誘導的中性粒細胞募集增加,嗜酸性粒細胞募集嚴重受損。這些結果證明Usp19缺陷在明礬誘導的腹膜炎模型中將抗炎反應轉換為促炎反應。
2. USP19可促進自噬介導的活性氧(ROS)清除,抑制NLRP3炎癥小體的激活
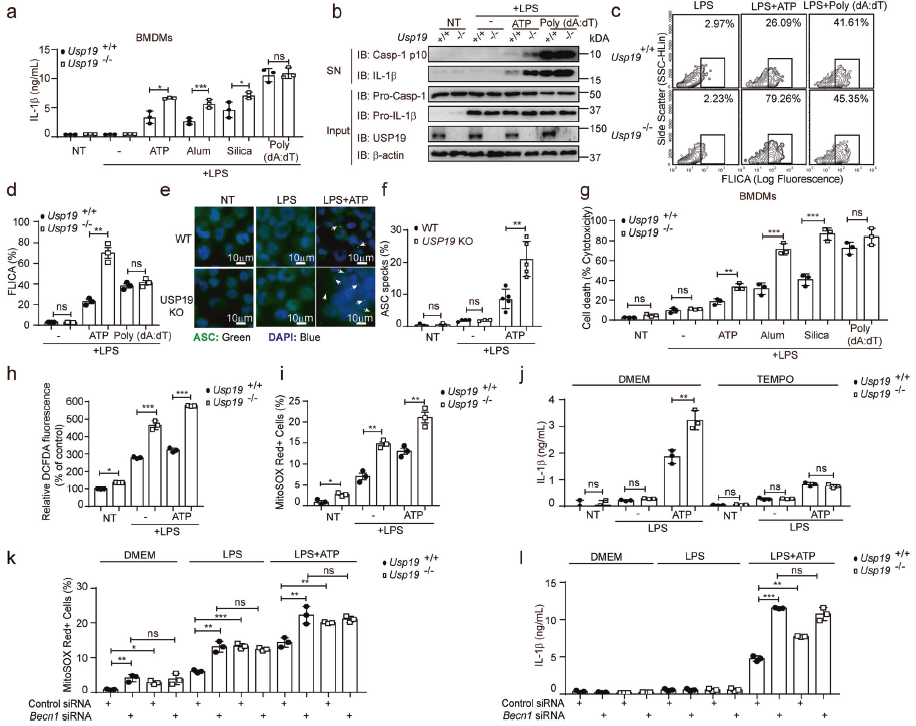
鑒于USP19在明礬誘導的腹膜炎模型中顯著抑制IL-1β的產生,推測USP19可能調節炎癥小體的激活。從Usp19-/-小鼠中分離出不同的細胞類型,發現在骨髓源性巨噬細胞(BMDMs)、骨髓源性樹突狀細胞(BMDC)和小鼠胚胎成纖維細胞(MEFs)中,Usp19缺失顯著增加ATP、明礬和二氧化硅刺激引起的IL-1β釋放,而AIM2炎癥小體激活劑誘導的IL-1β分泌保持不變。ATP處理的Usp19-/-小鼠BMDMs上清液中裂解的caspase-1增加。炎癥小體激活過程中,NLRP3通過PYD–PYD介導的相互作用觸發ASC斑點的形成。在USP19-KO THP-1細胞中,脂多糖LPS致敏和ATP激發的THP-1來源的巨噬細胞中ASC斑點形成在炎癥小體激活后顯著增強,在USP19-KO THP-1來源的巨噬細胞中敲低內源性NLRP3,發現NLRP3的耗竭消除了USP19-KO細胞對LPS和ATP的IL-1β上調反應。這些表明USP19缺陷特異性地增強NLRP3炎癥小體激活。
為確定USP19是否通過ROS的產生影響NLRP3炎癥小體的激活,檢測發現USP19缺乏增強了ROS的產生,線粒體靶向抗氧化劑處理巨噬細胞可消除Usp19-/-巨噬細胞中IL-1β分泌的增加。這些表明,USP19的耗竭通過上調線粒體ROS產生增強NLRP3炎癥小體激活。在缺乏Beclin-1的情況下,Usp19缺失導致的ROS上調降低,Beclin-1的缺失消除了USP19對IL-1β分泌的影響。這些表明,USP19/Beclin-1的缺失損害了自噬,導致ROS產生增加和隨后的NLRP3炎癥小體激活。
3. USP19對M2樣巨噬細胞極化至關重要
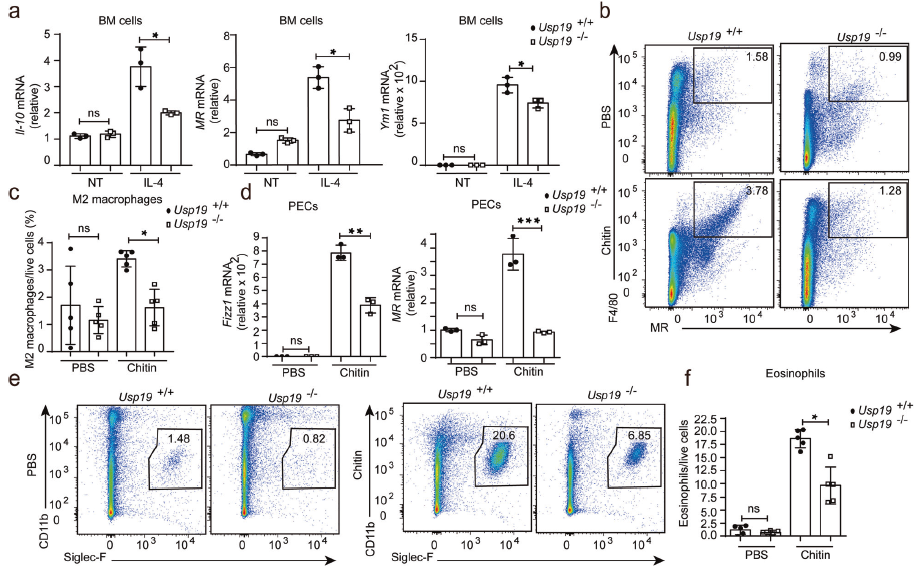
IL-4處理WT和Usp19-/-巨噬細胞,發現M2樣巨噬細胞相關標記物顯著降低。給小鼠腹腔注射甲殼素,發現從Usp19-/-小鼠獲得的腹腔中M2樣巨噬細胞比例降低,嗜酸性粒細胞數量也減少。這些表明USP19對甲殼素給藥后促進M2樣巨噬細胞極化至關重要。
4. USP19阻止IRF4被p62介導的選擇性自噬降解
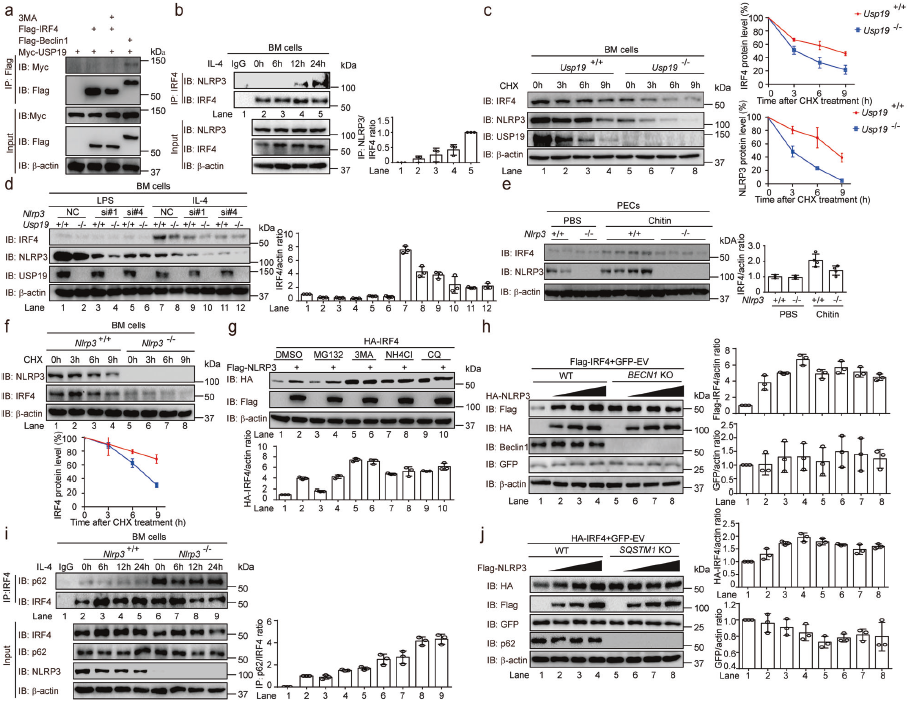
放線菌酮試驗發現USP19缺陷導致M2樣巨噬細胞的關鍵轉錄因子IRF4顯著降解,USP19的過表達增加了NLRP3穩定表達的人胚腎細胞中IRF4蛋白水平。自噬抑制劑、溶酶體抑制劑以及蛋白酶體抑制劑可完全抑制USP19介導的IRF4穩定作用。檢測IRF4與不同受體的相互作用,發現IRF4與p62特異性相互作用,USP19缺陷顯著增強p62和IRF4之間的關聯。在Usp19-/-BM細胞中重新表達了小鼠IRF4,恢復了IL-4誘導的Usp19-/-BM細胞中M2樣巨噬細胞極化。這些表明USP19通過阻止IRF4被p62介導的選擇性自噬降解,促進M2樣巨噬細胞極化。

5. USP19通過NLRP3穩定IRF4
為研究USP19如何阻止p62介導的IRF4降解。檢測到NLRP3缺陷降低了M2樣巨噬細胞的比例,NLRP3-/-BM細胞中小鼠NLRP3的再表達恢復IL-4誘導的M2樣巨噬細胞極化。IL-4刺激的BM細胞中,IRF4與NLRP3相關,并且IRF4的N末端對于IRF4–NLRP3結合至關重要。Usp19缺陷通過影響NLRP3和IRF4的降解速率而降低其蛋白水平。敲除NLRP3可顯著降低USP19誘導的IRF4蓄積。溶酶體抑制劑單獨穩定IRF4,加入NLRP3并不能進一步穩定IRF4。在自噬嚴重受損的BECN1-KO細胞中,NLRP3不能進一步增加IRF4蛋白水平。外源性NLRP3消除了IRF4–p62相互作用,NLRP3缺陷顯著增加了IL-4誘導的IRF4–p62結合。這些表明NLRP3通過阻斷IRF4–p62相互作用抑制IRF4降解。
6. USP19與NLRP3相互作用并穩定NLRP3
在BM細胞中,內源性USP19和NLRP3直接相關,但其相互作用因炎癥小體激活而降低。NLRP3 R260W突變體是NLRP3的一種活性形式,在沒有刺激的情況下直接觸發IL-1β分泌,結合USP19的能力下降,USP19不能穩定NLRP3 R260W突變體。在蛋白酶體抑制劑存在的情況下,USP19介導的NLRP3蓄積消失。USP19在K689切割NLRP3的多聚泛素鏈,抑制NLRP3的蛋白酶體降解。

總結:
1. USP19促進自噬,清除ROS,下調NLRP3炎癥小體激活。
2. USP19通過去除NLRP3的K689上的多聚泛素鏈,穩定炎癥小體中未摻入的NLRP3,促進NLRP3與IRF4的相互作用,從而阻止IRF4的p62依賴的選擇性自噬降解。
