






p53/microRNA-214/ULK1軸—糖尿病腎病治療的靶點(diǎn)
糖尿病腎病(DKD)中自噬調(diào)節(jié)失調(diào)已有報(bào)道,但其潛在機(jī)制及其致病作用仍不清楚。今天小編為大家介紹近期發(fā)表于影響因子11.864的雜志“The Journal of Clinical Investigation”的文獻(xiàn)“p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease”,帶大家了解自噬與DKD發(fā)展的機(jī)制。

在本文中,自噬在DKD模型和人類糖尿病腎臟中受到抑制。DKD的自噬損傷與ULK1的下調(diào)有關(guān),ULK1由糖尿病腎細(xì)胞和組織中miR-214的上調(diào)介導(dǎo)。從腎臟近端小管切除miR-214可防止糖尿病腎臟ULK1表達(dá)降低和自噬受損,從而減少腎臟肥大和蛋白尿。此外,p53的阻斷減弱了miR-214在DKD的誘導(dǎo),導(dǎo)致更高水平的ULK1和自噬,伴隨著DKD的改善。與非糖尿病樣本相比,糖尿病患者的腎活檢顯示p53和miR-214的誘導(dǎo),與ULK1和自噬的下調(diào)有關(guān)。我們還發(fā)現(xiàn)p53/miR-214與腎纖維化呈正相關(guān),但ULK1/LC3與糖尿病患者的腎纖維化呈負(fù)相關(guān)。
技術(shù)路線:
結(jié)果:
1) 糖尿病小鼠腎小管細(xì)胞自噬減少
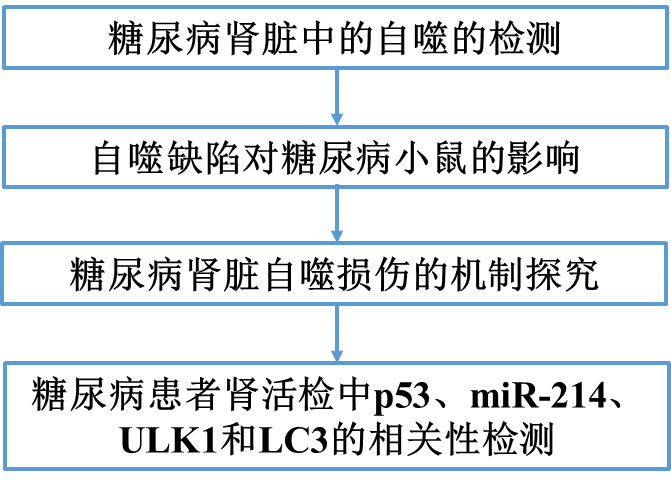
為了檢測(cè)糖尿病腎臟中的自噬,我們首先測(cè)試了Akita 小鼠,一種胰島素基因突變的1型糖尿病模型。Akita小鼠腎組織中LC3-I和LC3-II的水平明顯低于野生型小鼠(圖1A)。免疫組化染色顯示,與WT小鼠相比,Akita小鼠腎小管細(xì)胞胞漿中LC3陽性的斑點(diǎn)較少(圖1B)。電子顯微鏡還顯示,與野生型小鼠腎臟相比,Akita小鼠腎臟的腎小管細(xì)胞中自噬小泡較少(圖1C)。我們進(jìn)一步監(jiān)測(cè)了Akita小鼠糖尿病期間腎臟自噬變化的時(shí)間進(jìn)程。7周齡時(shí),Akita鼠和WT鼠的腎LC3表達(dá)均無差異(圖1D)。LC3在Akita小鼠腎臟中的表達(dá)在第9周較低(圖1E),但這種差異在第11周才具有統(tǒng)計(jì)學(xué)意義(圖1F),表明糖尿病患者腎臟自噬隨時(shí)間而減少。
我們還檢測(cè)了STZ誘導(dǎo)的C57BL/6小鼠糖尿病中的腎自噬。第11周,STZ處理的小鼠腎臟中LC3-I和LC3-II的表達(dá)水平明顯低于對(duì)照組小鼠(圖2A)。與未治療的對(duì)照組小鼠相比,這些小鼠的腎小管細(xì)胞中的LC3點(diǎn)也較少(圖2B)。
在糖尿病腎臟中觀察到的自噬減少可能是由于自噬體形成和/或自溶體降解的變化。因此,我們通過使用CAG-RFP-GFP-LC3-轉(zhuǎn)基因小鼠進(jìn)一步監(jiān)測(cè)自噬動(dòng)力學(xué),其中紅色RFP與綠色GFP熒光的點(diǎn)狀共定位指示自噬體,而沒有GFP信號(hào)的僅RFP點(diǎn)狀共定位指示自溶體。如圖2C所示,與對(duì)照動(dòng)物相比,STZ處理的小鼠腎小管細(xì)胞中的GFP- 和RFP-LC3 斑點(diǎn)明顯較少,表明糖尿病腎的腎小管整體自噬受損。

2)近端小管的自噬缺陷擴(kuò)大了糖尿病小鼠的腎臟肥大和組織損傷
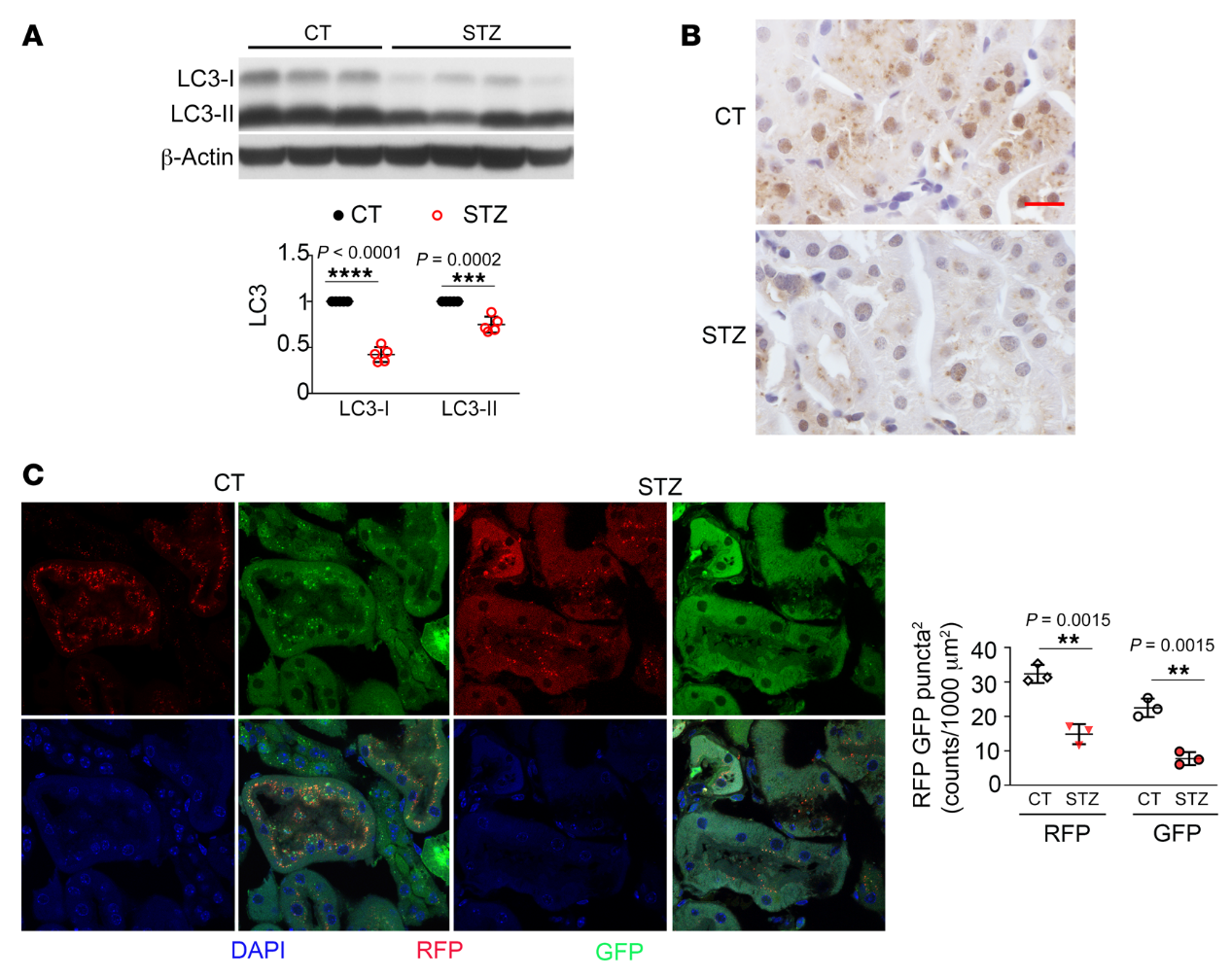
自噬是細(xì)胞分解代謝或降解的過程。因此,我們假設(shè)在糖尿病腎小管中觀察到的自噬損傷可能導(dǎo)致腎臟肥大,這是DKD病的早期發(fā)病特征。為了驗(yàn)證這一點(diǎn),我們首先確定了Akita小鼠腎臟近端小管自噬缺陷的影響。我們發(fā)現(xiàn)PT-Atg 7-/-Akita小鼠比PT-Atg7+/+ Akita小鼠具有更高的腎臟/體重比(圖3A),這表明當(dāng)腎小管自噬被抑制時(shí),糖尿病腎臟會(huì)發(fā)生更嚴(yán)重的肥大。腎臟組織學(xué)分析顯示,PT-Atg7-/-Akita小鼠的腎小管細(xì)胞嗜酸(圖3B)。此外,PT-Atg7-/- Akita小鼠顯示出腎小管損傷的其他跡象,包括腎小管細(xì)胞溶解和腎小管完整性喪失(圖3B)。然后我們檢查了巨噬細(xì)胞浸潤。如圖3C所示,我們?cè)诜翘悄虿T小鼠(不考慮Atg7表達(dá))和Atg7表達(dá)的Akita鼠的腎組織中觀察到很少的巨噬細(xì)胞。然而,PT-Atg7-/- Akita小鼠的腎間質(zhì)中有大量巨噬細(xì)胞浸潤,表明糖尿病期間近端小管的自噬缺陷促進(jìn)了腎臟炎癥。我們也用TUNEL染色檢查腎細(xì)胞死亡。我們?cè)赑T-Atg7+/+ WT腎臟中未檢測(cè)到TUNEL陽性細(xì)胞。在PT-Atg7–/–WT和PT-Atg7+/+Akita小鼠腎中觀察到少量TUNEL陽性細(xì)胞,但在PT-Atg7-/- Akita小鼠腎中發(fā)現(xiàn)更多的TUNEL陽性細(xì)胞(圖3D)。
3)腎小管自噬缺陷對(duì)DKD的長期影響。
用PAS染色,PT-Atg7-/- Akita小鼠比PT-Atg7+/+ Akita小鼠表現(xiàn)出更嚴(yán)重的腎小管損傷 (圖4A)。此外,我們觀察到PT-Atg7-/- Akita小鼠腎臟間質(zhì)明顯變寬(圖4A)。同樣,Masson’s trichrome染色檢測(cè)到PT-Atg7-/- WT和PT-Atg7+/+ Akita小鼠腎間質(zhì)中的邊緣膠原沉積,這在PT-Atg7-/- Akita小鼠中顯著增加(圖4B)。PT-Atg 7-/- Akita小鼠的腎間質(zhì)纖維連接蛋白染色也比其他組的小鼠多(圖4C)。從功能上來說,與WT小鼠相比,PT-Atg7+/+和PT-Atg7-/- Akita小鼠的尿白蛋白/肌酐比(ACR)更高,但PT-Atg 7-/- Akita小鼠的ACR明顯高于PT-Atg7+/+Akita小鼠(圖4D),這表明腎小管自噬缺乏會(huì)加重糖尿病患者的腎功能衰退。總之,這些結(jié)果表明,腎臟近端小管的自噬缺陷可能會(huì)加重糖尿病患者的腎臟肥大和組織損傷,從而促進(jìn)DKD病的進(jìn)展。

4)糖尿病腎臟和高糖孵育的腎小管細(xì)胞中ULK1的下調(diào)
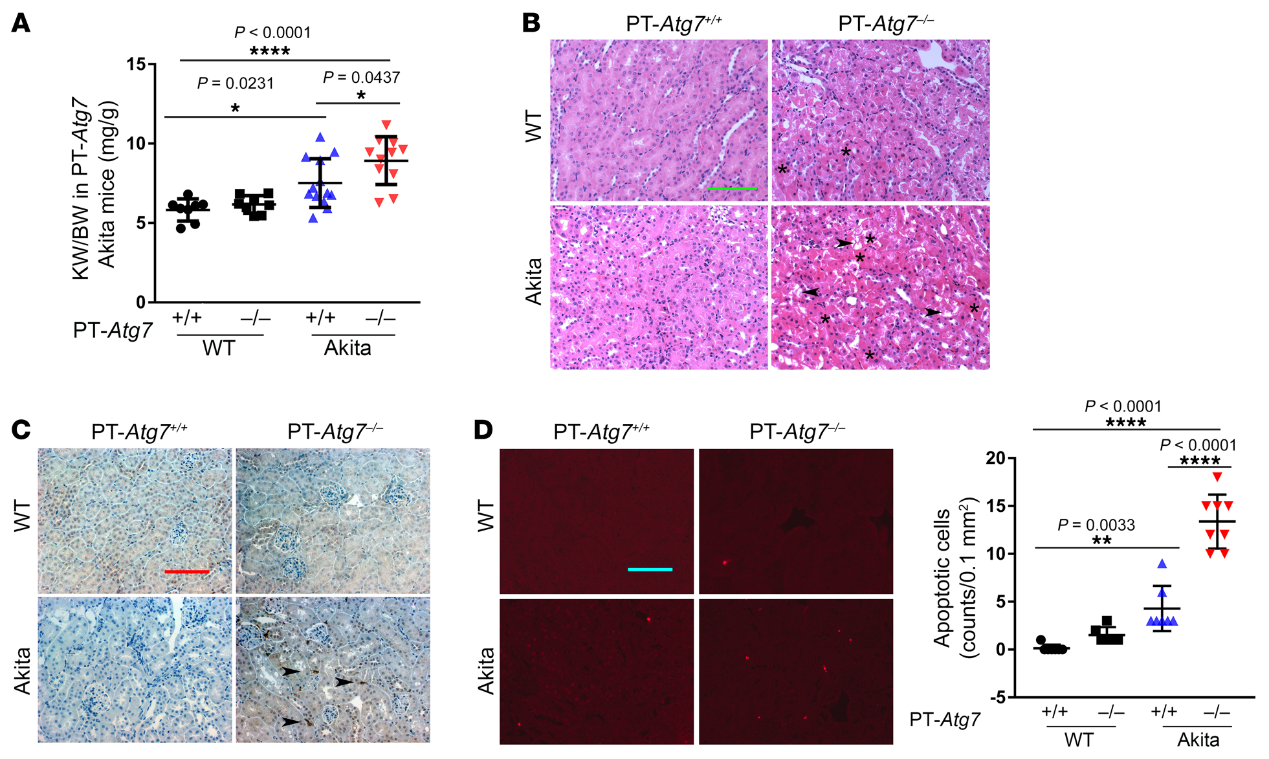
為了了解糖尿病腎臟自噬損傷的機(jī)制,我們首先通過免疫印跡法分析了Akita小鼠的關(guān)鍵自噬蛋白。我們發(fā)現(xiàn)糖尿病小鼠和它們的同窩仔在腎臟中表達(dá)相似水平的Beclin1和Atg5-Atg12復(fù)合物(圖5A)。IHC染色進(jìn)一步證實(shí)了糖尿病腎臟中ULK1的減少,尤其是在腎小管中(圖5B)。類似地,在STZ誘導(dǎo)的糖尿病小鼠中,ULK1的腎表達(dá)減少(圖5,C和D)。然而,STZ治療沒有改變小鼠腎臟中Atg7、Atg5或Beclin1的表達(dá)(圖5C)。因此,糖尿病腎臟中一個(gè)顯著的分子變化是ULK1的下調(diào),這可能介導(dǎo)糖尿病中的自噬損傷。
5)在糖尿病腎臟中誘導(dǎo)miR-214抑制ULK1的自噬損傷、腎臟肥大和DKD
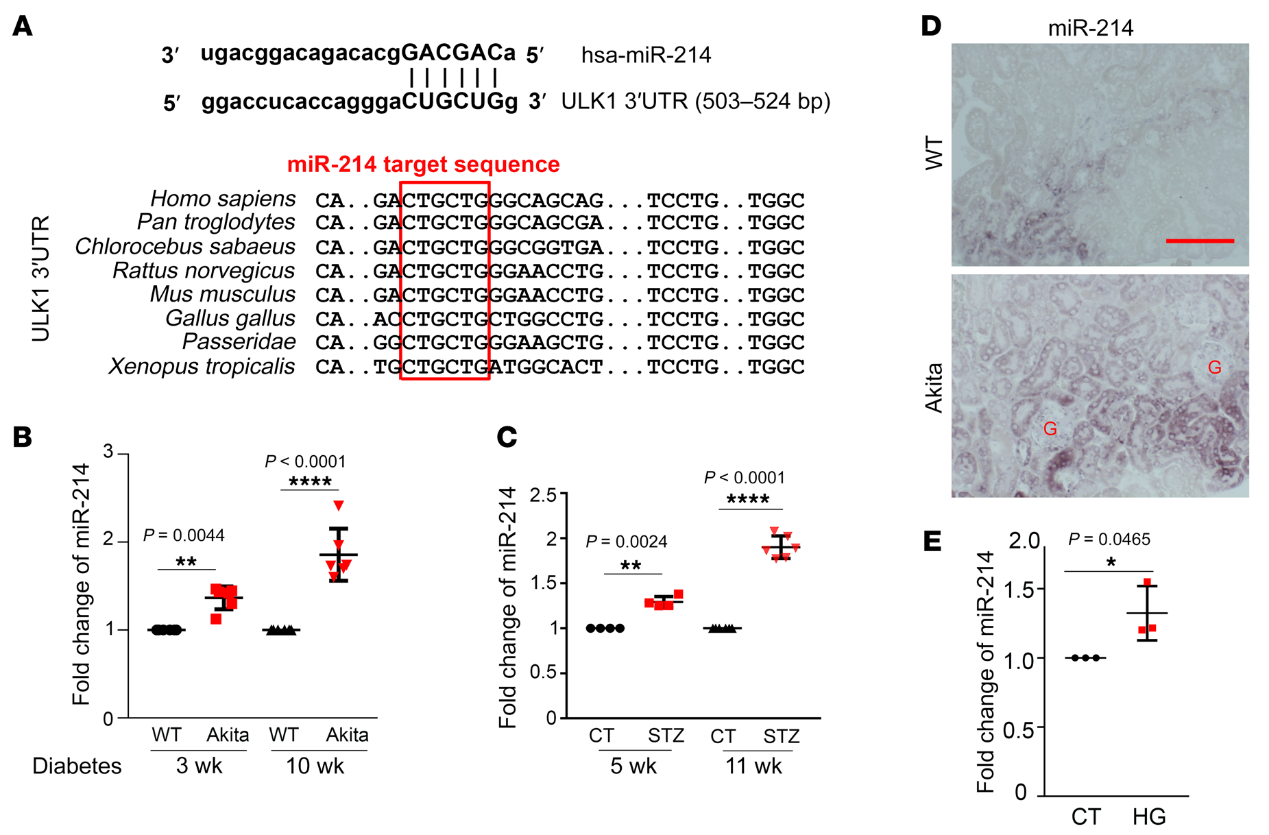
為了識(shí)別特定的microRNAs,我們首先使用miRanda microRNA目標(biāo)預(yù)測(cè)數(shù)據(jù)庫(http:/ /www.microrna. org)來預(yù)測(cè)可能以ULK1為目標(biāo)的microRNAs。然后我們集中研究了那些與糖尿病和腎臟疾病有關(guān)的microRNAs。生物信息學(xué)分析表明miR-214是一種潛在的microRNAs,可以靶向糖尿病腎臟中的ULK1。我們?cè)趶臒釒ё傅饺祟惖母鞣N動(dòng)物物種中鑒定了ULK1基因3’-UTR中的一個(gè)保守的miR-214靶向位點(diǎn)(圖6A)。我們進(jìn)一步檢測(cè)到Akita小鼠和STZ誘導(dǎo)的糖尿病小鼠腎臟中miR-214的表達(dá)逐漸增加(圖6,B和C)。使用ISH,我們進(jìn)一步定位了糖尿病腎臟中miR-214的誘導(dǎo),主要是在近端小管,而不是在腎小球(圖6D),與非糖尿病WT組織相比,Akita小鼠腎臟中miR-214陽性管狀細(xì)胞的數(shù)量顯著增加。在體外RPTCs中,HG誘導(dǎo)miR214增加30%(圖6E)。總之,這些結(jié)果證明了糖尿病期間腎小管細(xì)胞中miR-214的誘導(dǎo),伴隨著ULK1的下調(diào)。
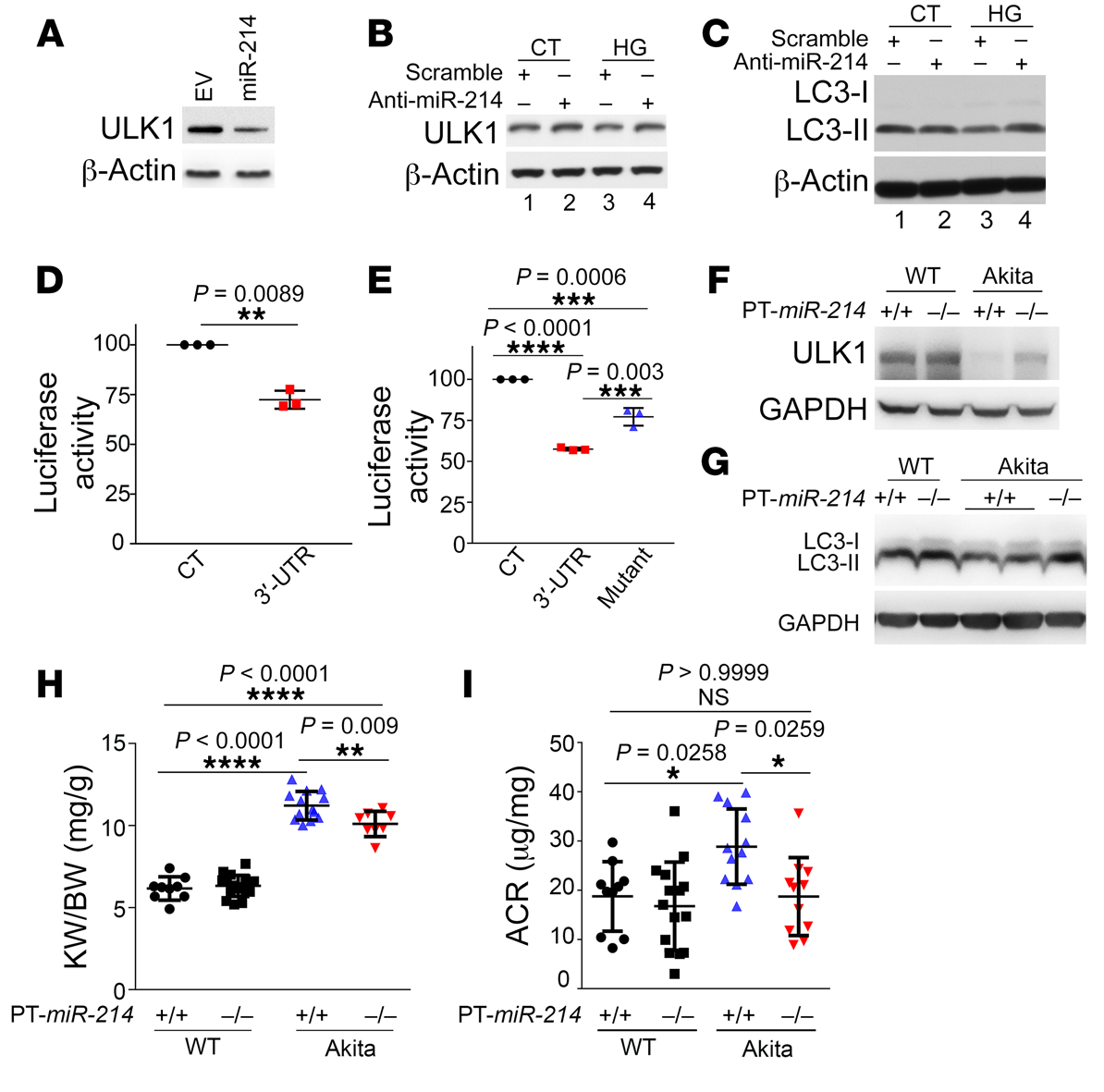
為了確定ULK1是否是miR-214的真正靶標(biāo),我們首先將miR-214轉(zhuǎn)染到人胚胎腎293 (HEK293)細(xì)胞中,這導(dǎo)致ULK1表達(dá)的顯著降低(圖7A)。相反, miR-214的抑制增強(qiáng)ULK1表達(dá)(圖7B)。此外,抗miR214可防止HG誘導(dǎo)的ULK1(圖7B)和LC3-II(圖7C)表達(dá)的降低。我們通過熒光素酶報(bào)告分析,進(jìn)一步確定ULK1是否是miR-214的直接靶點(diǎn)。如圖7D所示,共轉(zhuǎn)染miR-214降低了熒光素酶-ULK 1 3’-UTR轉(zhuǎn)染細(xì)胞中的熒光素酶表達(dá)。值得注意的是,HG孵育也抑制了轉(zhuǎn)染細(xì)胞中熒光素酶的表達(dá),當(dāng)miR-214的靶向序列在熒光素酶-ULK1 3’-UTR中突變時(shí),這種作用部分減弱(圖7E)。總之,這些數(shù)據(jù)表明ULK1是miR-214的直接靶標(biāo),miR-214可能抑制糖尿病中ULK1的表達(dá)。
為了證實(shí)anti–miR-214的結(jié)果,并進(jìn)一步闡明miR-214在糖尿病腎臟中的致病作用,我們建立了近端小管特異性miR-214-敲除(PT-miR-214-/-)小鼠模型。PT–miR-214+/+ Akita小鼠腎臟中ULK1和LC3-II的表達(dá)降低,這兩種蛋白在PT–MiR-214-/- Akita小鼠中被部分阻止 (圖7,F(xiàn)和G)。此外,PT–miR-214-/-Akita小鼠的腎臟和腎臟重量/體重比小于PT–miR-214+/+ Akita小鼠,這表明miR-214有助于糖尿病患者的腎臟肥大(圖7H)。值得注意的是,PT–miR-214-/- Akita小鼠的腎功能也明顯更好(圖7I)。
6)P53介導(dǎo)糖尿病腎臟中miR-214誘導(dǎo)的自噬損傷、腎臟肥大和DKD
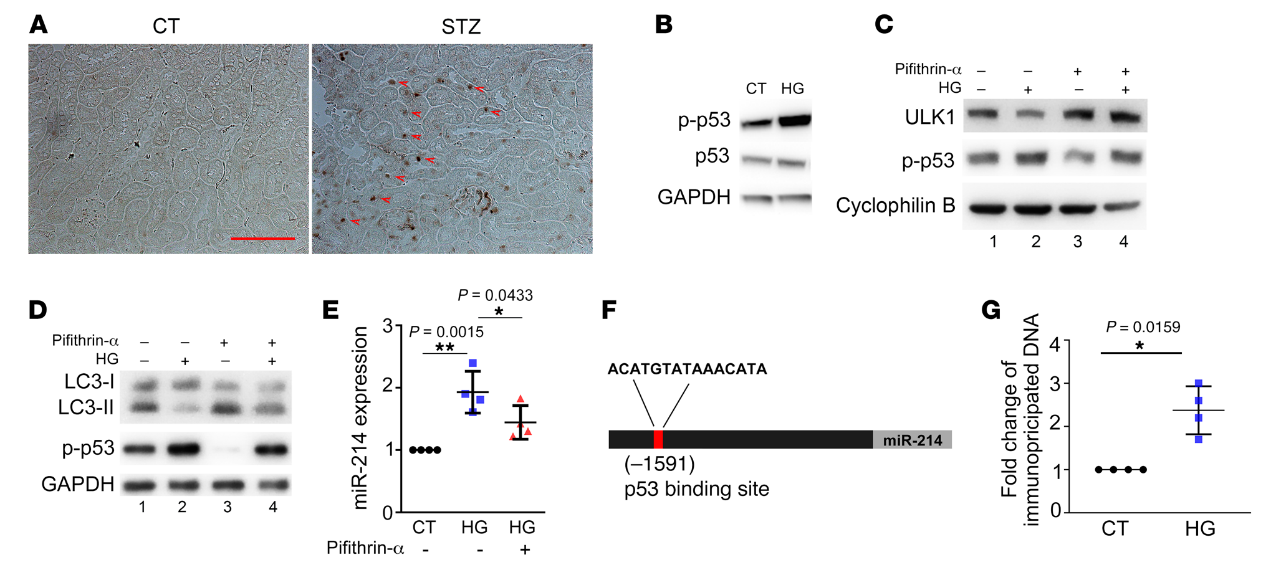
miR-214在DKD是如何誘導(dǎo)的?考慮到這個(gè)問題,我們篩選了幾種可能的上游轉(zhuǎn)錄因子,其中p53在Akita和STZ治療的小鼠腎臟中顯著上調(diào)和激活(圖8A)。在體外,RPTCs的HG孵育也誘導(dǎo)p53活化(圖8B)。Pifithrin-α是p53的一種藥理學(xué)抑制劑,可防止HG處理的RPTCs中ULK1和LC3-II表達(dá)的降低(圖8,C和D)。值得注意的是,pifithrin-α還降低了HG處理細(xì)胞中miR-214的誘導(dǎo)(圖8E)。這些結(jié)果表明p53可能介導(dǎo)腎小管細(xì)胞中miR-214的誘導(dǎo)以抑制糖尿病中的ULK1和自噬。
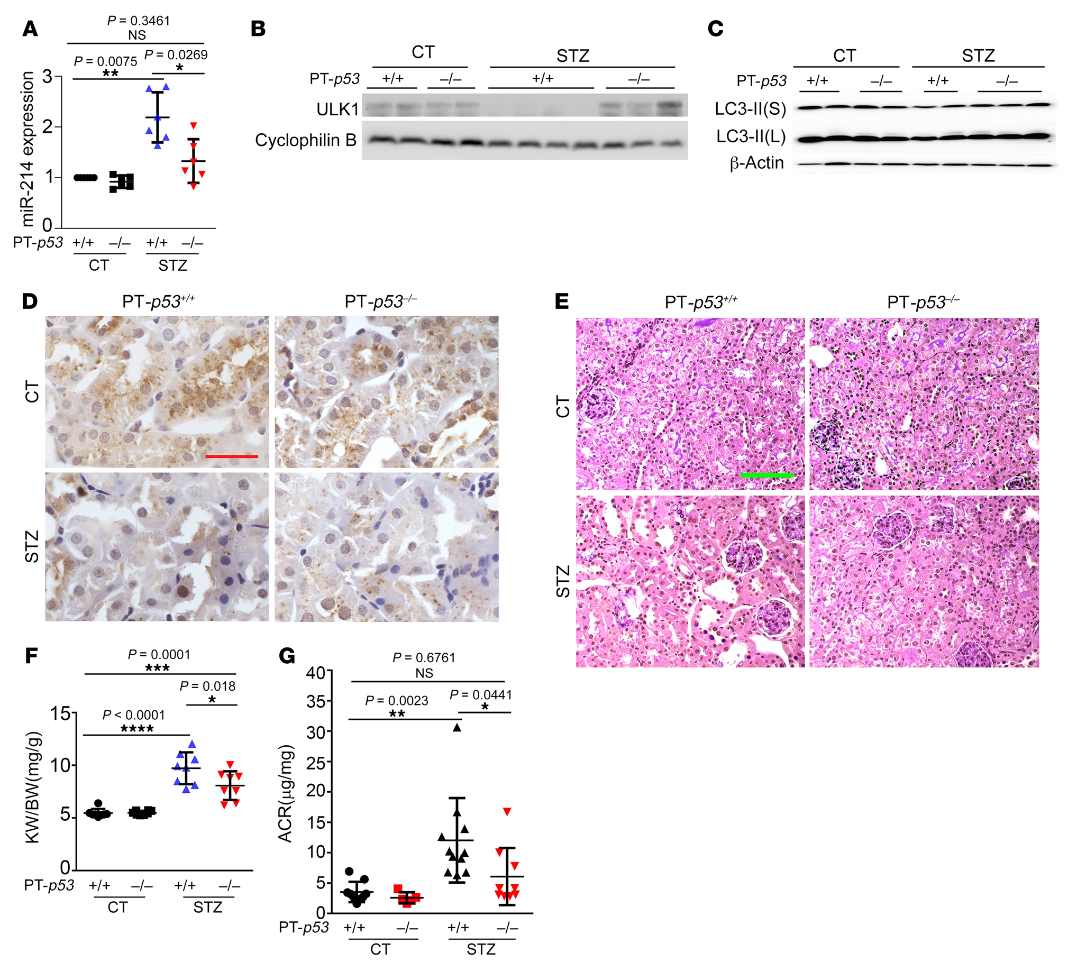
然后,我們使用JASPAR數(shù)據(jù)庫(http:/ /jaspar.genereg.net/)分析miR-214基因的轉(zhuǎn)錄因子結(jié)合譜,并在其啟動(dòng)子區(qū)鑒定出一個(gè)假定的p53結(jié)合位點(diǎn)(圖8F)。為了驗(yàn)證p53與該位點(diǎn)的結(jié)合,我們進(jìn)行了ChIP分析以檢測(cè)抗p53免疫沉淀中的結(jié)合位點(diǎn)序列。如圖8G所示,HG孵育誘導(dǎo)p53與miR-214基因啟動(dòng)子序列的結(jié)合增加了2.3倍,該序列具有假定的結(jié)合位點(diǎn)。為了闡明p53是否在體內(nèi)調(diào)節(jié)miR-214,我們使用了我們以前工作中的近端小管特異性p53敲除(PT-p53–/–)小鼠模型。PT-p53-/-小鼠和它們的PT-p53+/+同窩小鼠用STZ治療以誘發(fā)糖尿病。經(jīng)STZ治療后,PT-p53+/+小鼠腎臟中miR-214的誘導(dǎo)作用顯著,而PT-p53-/-小鼠中miR-214的誘導(dǎo)作用減弱(圖9A)。與此同時(shí),糖尿病相關(guān)的ULK1和LC3-II表達(dá)的降低在PT-p53-/-小鼠中被抑制(圖9,B和C)。在STZ誘導(dǎo)糖尿病后,PT-p53-/-腎在腎小管中也比PT-p53+/+腎有更多的LC3陽性點(diǎn)或自噬體(圖9D)。在PAS染色中,糖尿病性PT-p53+/+小鼠表現(xiàn)出明顯的腎臟組織病理學(xué)變化,包括腎小管擴(kuò)張、刷狀緣喪失和腎小管萎縮,這些在糖尿病性PT-p53-/-小鼠中顯著減少(圖9E)。糖尿病期間腎臟重量和腎臟重量/體重比增加,但PT-p53-/-小鼠的增加被部分抑制,表明這些動(dòng)物的腎臟肥大減少(圖9F)。值得注意的是,在STZ誘導(dǎo)的糖尿病中,PT-p53-/-小鼠的ACR顯著低于PT-p53+/+小鼠(圖9G),表明腎功能更好。綜上所述,這些結(jié)果表明糖尿病中p53激活以誘導(dǎo)miR-214,miR-214隨后抑制ULK1的表達(dá),導(dǎo)致自噬損傷、腎肥大和DKD。
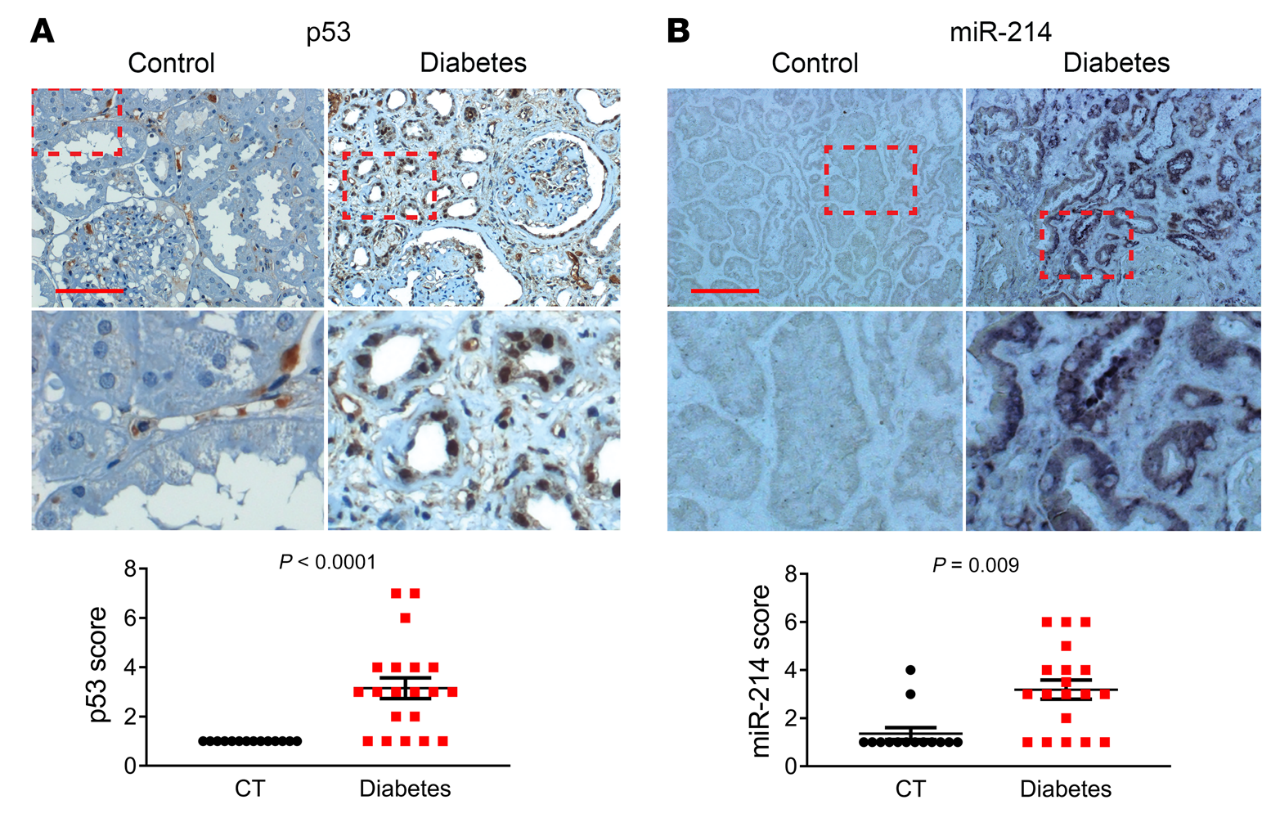
7)糖尿病患者腎活檢中p53、miR-214、ULK1和LC3的相關(guān)性
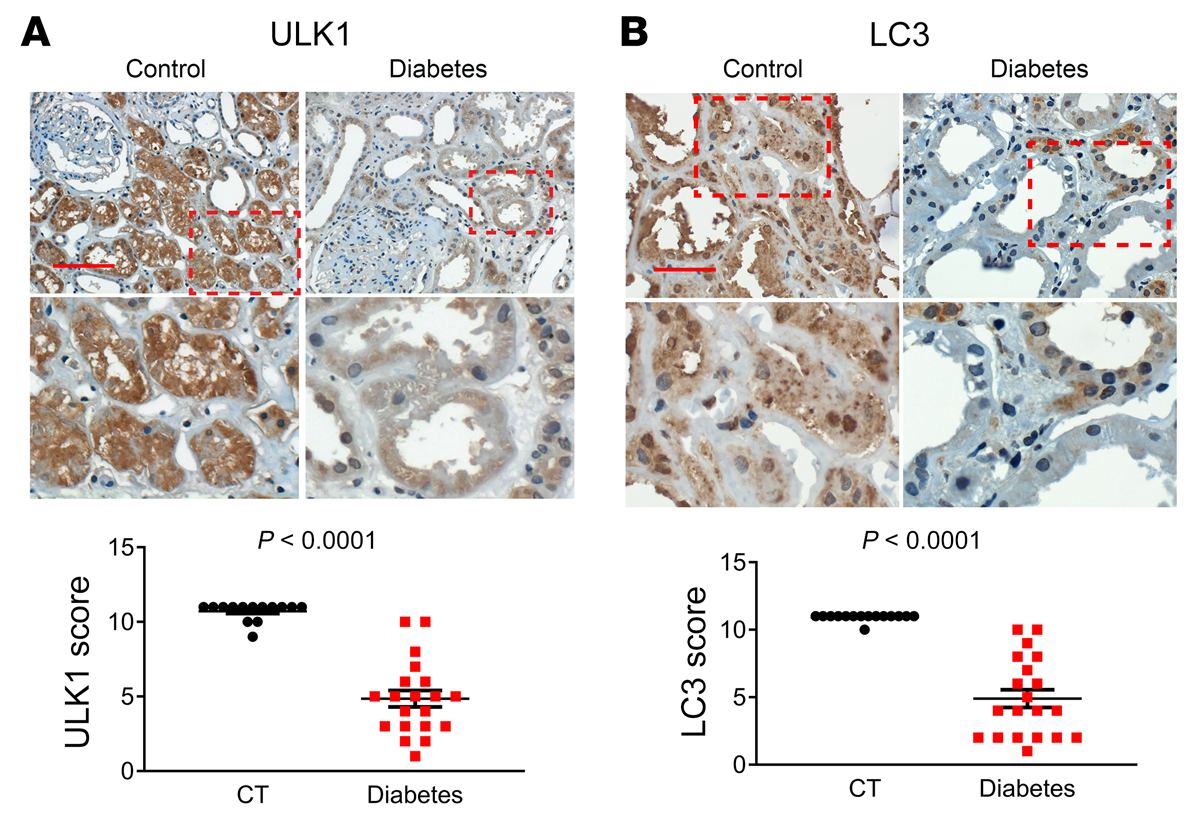
我們的動(dòng)物和細(xì)胞模型結(jié)果揭示了p53/miR-214/ULK1途徑,該途徑導(dǎo)致自噬缺陷、腎臟肥大和糖尿病腎功能下降。為了確定這些發(fā)現(xiàn)的臨床相關(guān)性,我們檢測(cè)了糖尿病患者腎活檢中p53、miR-214、ULK1和LC3的表達(dá)。在IHC分析中,沒有一個(gè)對(duì)照腎活檢標(biāo)本p53染色陽性,而來自糖尿病患者的20個(gè)腎活檢標(biāo)本中有15個(gè)顯示p53染色(圖10A)。值得注意的是,在糖尿病腎臟樣本中,p53主要在擴(kuò)張腎小管的細(xì)胞核中表達(dá)。ISH顯示,14例對(duì)照腎活檢中只有2例miR-214染色陽性,而糖尿病患者20例腎活檢中有15例陽性(圖10B)。有趣的是,miR-214在腎小管中以p53的形式存在,但主要在細(xì)胞質(zhì)中。相反,對(duì)照腎活檢中的大多數(shù)腎小管對(duì)ULK1染色強(qiáng)烈,而糖尿病腎活檢中的許多腎小管對(duì)ULK1染色較低(圖11A)。與對(duì)照活檢相比,糖尿病患者的腎活檢也顯示明顯較少的LC3點(diǎn)或自噬體(圖11B)。此外,線性回歸分析顯示糖尿病腎活檢中p53和miR-214表達(dá)之間(圖12A)以及ULK1和LC3表達(dá)之間(圖12B)存在顯著正相關(guān)。相比之下,我們注意到miR-214和ULK1的表達(dá)之間(圖12C)以及p53和ULK1的表達(dá)之間(圖12D)存在顯著的負(fù)相關(guān)。這些結(jié)果提供了進(jìn)一步支持p53/miR-214/ULK1軸在糖尿病自噬損傷中的作用,并建立其臨床相關(guān)性。

結(jié)論:我們?cè)贒KD的實(shí)驗(yàn)?zāi)P秃吞悄虿』颊叩哪I臟中都證實(shí)了腎小管細(xì)胞的自噬功能障礙。自噬損傷有助于腎臟肥厚,相關(guān)病理,以及DKD的進(jìn)展。機(jī)制上,我們發(fā)現(xiàn)p53誘導(dǎo)了DKD中的miR-214,抑制了自噬啟動(dòng)的關(guān)鍵蛋白激酶ULK1,導(dǎo)致自噬損傷。