






MTOR信號調節應激誘導的突變,促進癌癥的適應性進化
近日澳大利亞加文醫學研究所金霍恩癌癥中心David M. Thomas課題組發現,非遺傳毒性藥物選擇下的應激誘導突變(stress-induced mutagenesis,SIM),自相矛盾地以競爭的內在適應性成本提高腫瘤細胞適應性,表明了傳統的靶向癌癥治療與靶向DNA修復機制的藥物相結合,可以降低腫瘤耐藥性,可能是更有效的腫瘤治療策略。相關論文“MTOR signaling orchestrates stress-induced mutagenesis, facilitating adaptive evolution in cancer.”于2020年9月份發表在《科學》雜志上。
技術路線:

一、靶向抗癌療法的選擇導致了遺傳多樣性

為了了解暴露于非直接基因毒性療法下人類癌細胞中DNA損傷積累的情況,作者把磷酸化組蛋白H2AX(γ-H2AX)作為DNA雙鏈斷裂(DNA-DSBs)的標記物,并對不同化療和放療組的初發癌患者的樣本進行治療前后的檢測。結果顯示,在前列腺癌患者經雄激素剝奪治療后,乳腺癌患者經芳香化酶抑制劑來曲唑治療后,黑色素瘤患者經BRAF抑制劑達布拉芬尼或維穆拉芬尼治療后,胃腸道間質瘤患者使用KIT抑制劑伊馬替尼治療后,DNA損傷水平均出現明顯增加(fig1. a)。在CDK4/6抑制劑palbociclib和表皮生長因子受體(EGFR)抑制劑erlotinib治療的人源性異種移植(PDX)胰腺癌模型中DNA-DSBs亦出現增加,提示DNA損傷增加是非毒性治療的人類腫瘤的復發特征。隨后,作者將多種人腫瘤細胞系暴露于接近致死劑量的非毒性藥物下,發現每100000個腫瘤細胞仍能產生1~20個耐藥群落(fig1.b)。同時發現在藥物處理早期所有研究模型均顯示DNA-DSBs水平上升(fig1.c),但在隨后的培養過程中DNA損傷水平逐漸回歸到基線。
隨后,作者對94T778人脂肪肉瘤和SKMEL28人黑色素瘤細胞株經藥物處理后形成的單細胞克隆群體進行了全基因組測序。結果顯示,兩個細胞系的抗性群體中單核苷酸變異(SNV)率均顯著升高(fig1.e),而其加速進化的基因組與癌癥治療瓶頸期的基因組信息一致(fig1.d)。同時在藥物處理早期,腫瘤細胞因SIM導致克隆形成能力出現明顯下降(fig1.f)。
綜上所述,該研究在非直接基因毒性療法早期,人類癌細胞中DNA損傷水平出現暫時性的升高,并且SIM會在一定程度上降低腫瘤細胞的增殖能力。
二、全基因組RNAi篩選鑒定MTOR為癌癥中常見的進化電容器

對94T778細胞系進行了全基因組功能篩選,最終發現MTOR在提高腫瘤細胞適應性中發揮重要作用(fig2.a-b)。
MTOR信號通路在人類組織中表達廣泛,是一種進化上保守的環境和內源性應激傳感器。作者發現在無藥物處理時,沉默MTOR 會降低腫瘤細胞的克隆能力;而在藥物作用于腫瘤細胞時,MTOR的沉默反而會提高腫瘤細胞的克隆能力(fig2.c)。
在胰腺癌PDX模型中亦證實,抑制MTOR會加速palbociclib耐藥性的出現(fig2.d),且在腫瘤細胞進化早期MTOR及p-70-S6K磷酸化水平顯著降低,而在適應后期恢復至基礎水平。
三、抑制MTOR通過抑制準確的DNA修復來培養適應性突變
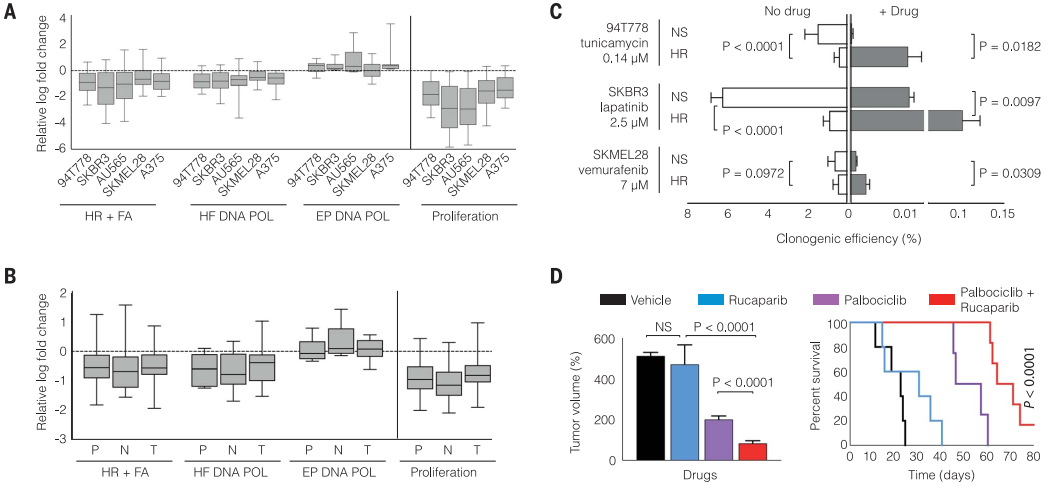
對94T778親本細胞系及MTOR沉默的單細胞克隆群進行了全基因組測序,發現MTOR沉默會抑制了同源重組(HR)信號通路的表達,并選擇性抑制了高保真DNA多聚酶活性(fig3.a)。
細胞系轉錄組數據庫LINCS L1000顯示,多種腫瘤細胞系在不同MTOR抑制劑處理下均出現細胞增殖能力的下降及DNA精確修復信號通路的抑制(fig3.b)。
靶向HR基因的shRNA處理94T778、SKBR3和SKMEL28細胞,結果顯示干擾HR后在無藥物處理的情況下細胞克隆能力下降,在藥物處理下細胞增殖能力卻增值(fig3.c)。
將palbociclib與rucaparib(聚腺苷二磷酸核糖聚合酶抑制劑)聯用以治療胰腺癌PDX模型,與單獨使用任何一種藥物相比,聯合療法增強了抗腫瘤作用(fig3.d)。
四、支持SIM在人類癌癥中的保守機制
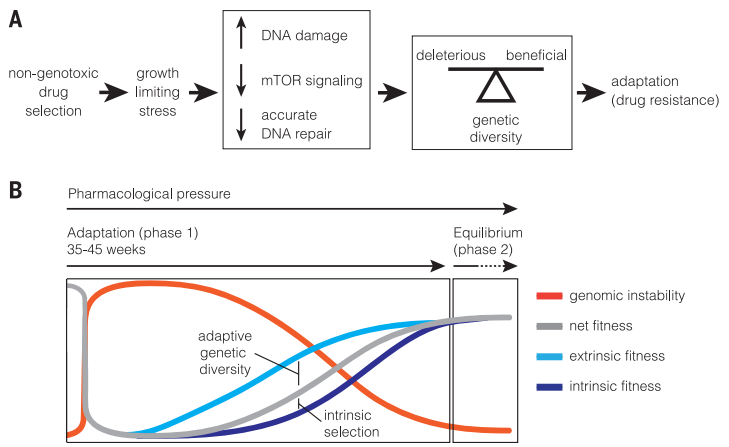
MTOR介導的SIM通過抑制DNA的精確修復機制而減緩細胞增殖并提高基因組不穩定性,從而增強遺傳多樣性并促進腫瘤細胞的耐藥性(fig4.a)。
因此耐藥可能是兩個獨立但相關機制的復合結果,而不是一個簡單的過程。最初MTOR介導的SIM增加了腫瘤細胞的遺傳多樣性,在藥物、輻照等選擇下,一些含有抗性基因的細胞得以生存,隨后細胞內通過SIM的標準化和穩定抗性基因組結構的建立而形成了對新環境的適應性(fig4.b)。