






Trib1/Hoxa9/Erg軸在急性髓系白血病進展中的作用
急性髓系白血病(AML)是成年人最常見的急性白血病,目前治療仍以化療為主,但有70%左右獲得緩解的患者最終復發并演變為難治性白血病,導致治療失敗而死亡。轉錄調控異常是AML常見的分子特征。在AML中,造血轉錄因子常被認定為癌基因或抑癌基因。Hox蛋白是關鍵的轉錄因子,Hoxa9調控的轉錄程序參與AML的MLL融合、MOZ-TIF2、NUP98-NSD1、NPM1c或ASXL1融合/突變。這些突變上調了HOXA9的mRNA表達,并揭示了HOXA9過表達對小鼠造血細胞永生化的意義。然而,僅過表達Hoxa9并不足以發展成全面的AML,需要激活額外的輔助因子和/或合作通路。研究表明,多種信號通路與Hoxa9調控的轉錄程序相互作用,額外的分子機制可能影響Hoxa9驅動的增強子修飾。最近,日本癌癥研究基金會的Takuro Nakamura教授及其團隊發現Trib1在髓系白血病發生中調控染色質和Hoxa9驅動轉錄的新機制,相關研究以“Trib1 promotes acute myeloid leukemia progression by modulating the transcriptional programs of Hoxa9”為題發表在Blood雜志上,雜志影響因子為17.543。
研究思路:

結果:
1.Trib1過表達對Hoxa9誘導髓系白血病生成的影響
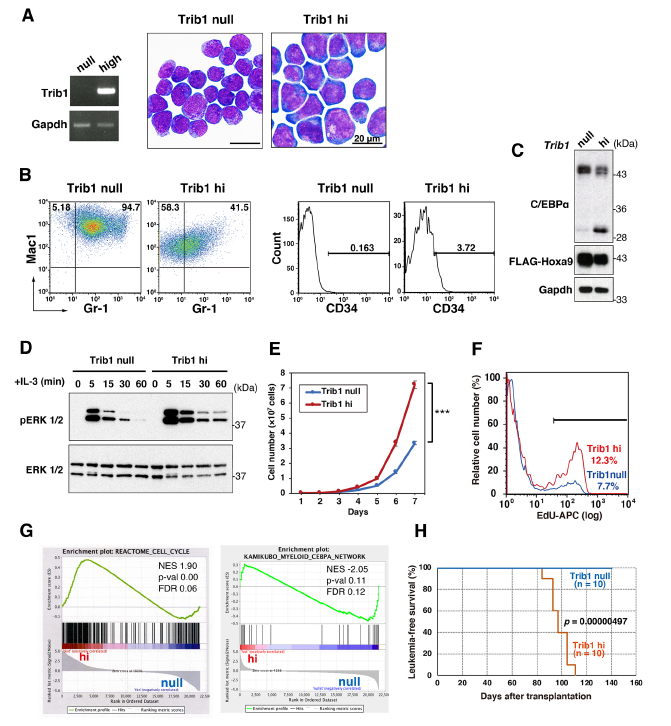
作者使用逆轉錄病毒載體pMYs-IRES-GFP將Hoxa9和/或Trib1導入Trib1敲除小鼠的骨髓細胞,建立了不朽的骨髓細胞系,表達(Trib1 hi)或不表達(Trib1 null)Trib1的Hoxa9細胞表現出未成熟的髓系形態(A)。兩種細胞類型Mac1和Gr1均呈陽性,而Trib1 hi細胞中Mac1和Gr1表達水平下降,CD34表達升高,提示Trib1 hi細胞較Trib1 null細胞具有更多未成熟特征(B)。Trib1 hi細胞中,C/EBPα p42而不是p30被發現顯著降低,ERK磷酸化增強,IL-3刺激后延長(C,D),且與Trib1 null相比,Trib1 hi細胞也顯示出更高的增殖率和EdU摻入率,暗示Trib1誘導細胞周期的增加(E,F)。基因微陣列分析Trib1 hi/null細胞發現Trib1 hi特異性富集細胞周期以及C/EBP通路(G)。骨髓移植實驗發現只有Trib1 hi細胞才能在體內誘導白血病發生(H)。這些結果表明Trib1過表達促進了Hoxa9誘導的白血病發生。
2.AML中Hoxa9和C/EBPα的DNA結合特性
Trib1 null和hi細胞中,Hoxa9的DNA結合峰的總體分布沒有顯著差異,其中39%和27%的DNA結合峰分別位于距離轉錄起始位點(TSS) 2 kb以內(A)。MEME分析發現Hoxa9 (FLAG)和C/EBP結合峰顯示了Hoxa9,C/EBPα和Meis1顯著的濃度,暗示Hoxa9和C/EBP在染色質水平上是接近的(B)。在Trib1缺失細胞中,Hoxa9和C/EBP的DNA結合出現了頻繁的關聯,且Hoxa9和C/EBP的結合峰有60.5%的重疊(C)。通過比較組蛋白H3K27ac和Hoxa9結合的分布。作者發現,在Trib1 null和hi細胞中,Hoxa9和H3K27ac峰經常同時出現(D),而在Trib1 null和hi細胞中,H3K27ac的整體沉積并沒有太大差異。在Trib1 hi和null細胞的啟動子區、基因內區和基因間區檢測到Hoxa9、C/EBP和H3K27ac的共沉積,此外,通過ChIP-seq檢測了C/EBP在Trib1 hi細胞中的DNA結合,發現由于p42的降解而富集了p30亞型,且41.1%的C/EBP結合位點在hi和null細胞之間重疊,暗示p30可能在hi細胞中發揮不同的作用(E)。

3.Trib1能夠調節Hoxa9誘導AML中的超級增強子
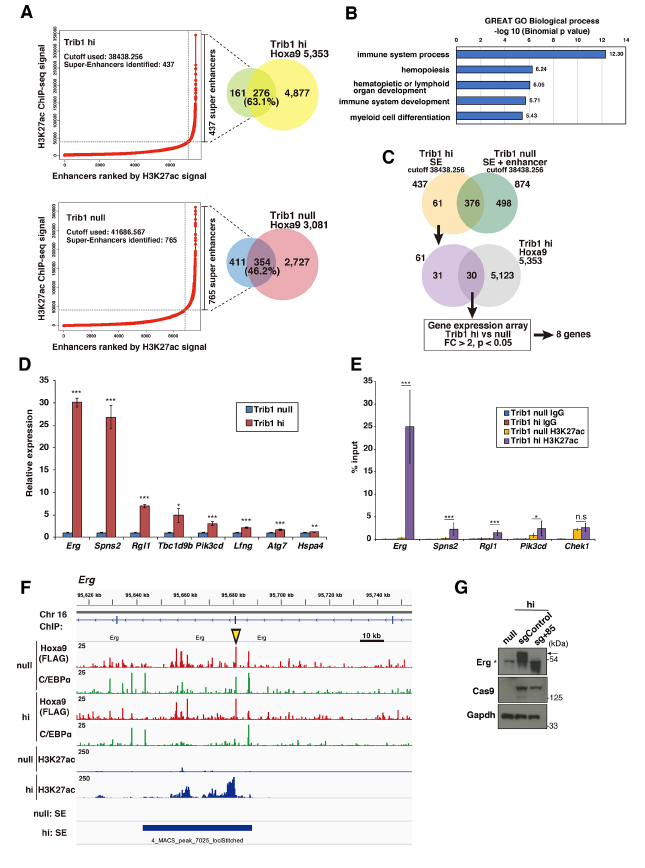
作者比較了Trib1 null和hi細胞間的超級增強子譜,發現437個和765個超級增強子分別與Trib1 null和hi細胞中63.1%和46.2%的Hoxa9結合位點相關(A)。通過對Trib1 hi或null特異性超級增強子的生物學過程通路的系統分析,我們確定了Trib1 hi細胞中的免疫系統、造血和骨髓細胞分化通路,提示Trib1 hi特異性超級增強子在造血系統中發揮重要作用(B)。作者識別由Trib1和Hoxa9調控修飾超級增強子活性的重要靶點,選擇了61個Trib1 hi特異性超級增強子,其中8個基因在Trib1 hi細胞中表達上調,并通過Trib1過表達鑒定為顯著修飾基因(C)。定量RT-PCR驗證這8個基因的mRNA表達水平顯著升高(D)。ChIP-qPCR顯示,Trib1 hi細胞中Erg、Spns2、Rgl1和Pik3cd基因座的H3K27Ac沉積顯著增加(E),且在這些基因中,包括+85干細胞增強子在內的Erg位點,可以觀察到Trib1 hi和null細胞之間H3K27Ac的顯著差異(F)。采用CRISPR/ cas9介導靶向Erg +85增強子,50 bp的純合缺失(包括增強子中一個假定的Hoxa9結合位點)消除了Erg的上調(G),表明Hoxa9在+85增強子上結合上調了Erg。
4.Trib1降解C/EBP對增強子和Hoxa9靶基因的作用
在MEK1抑制劑U0126處理的Trib1 hi細胞中,發現超級增強子在Erg、Spns2和Pik3cd位點的H3K27Ac積累受到抑制(A),然而,mRNA表達的下調是邊際性的(B)。Trib1 null細胞的Cebpa沉默后, Erg、Spns2、Rgl1和Pik3cd超級增強子的H3K27Ac信號顯著增加(C),且這四個基因的表達顯著上調(D)。通過將編碼全長(p42和p30)或N端截斷(僅p30)蛋白的人CEBPA cDNAs引入CEBPA沉默的Trib1 null細胞中,發現p42部分恢復了Erg mRNA和蛋白的表達以及組蛋白H3K27Ac的積累,而p30的表達沒有恢復(E)。綜上所述,Trib1對C/EBP p42的降解是某些Hoxa9結合位點上增強子修飾的主要驅動力,而MEK/ERK的增強對這一事件略有貢獻。

5.Erg是Trib1作用于Hoxa9的重要靶點
ChIP-seq分析顯示Trib1過表達上調了Erg水平,且Erg靶基因調控所需的Erg蛋白p55亞型的表達在Trib1 hi細胞中上調(A)。研究發現,與Trib1 hi細胞相比,表達Erg的Trib1 null細胞的增殖能力增強。相反,shRNA介導Erg沉默的Trib1 hi細胞顯示生長抑制(C),而CRISPR/ cas9介導Erg沉默進一步顯示Trib1 hi細胞的生長抑制等同于Trib1 null細胞(D)。此外,Erg缺失完全消除了體內Trib1 hi細胞的白血病發展(E),盡管在Trib1 null細胞中Erg過表達不會發展為白血病,這表明在與Hoxa9表達相關的Trib1增強AML中,Erg上調是必需的,但不足以促進體內白血病的發生。

6.BRD4抑制劑抑制Hoxa9誘導的白血病生長
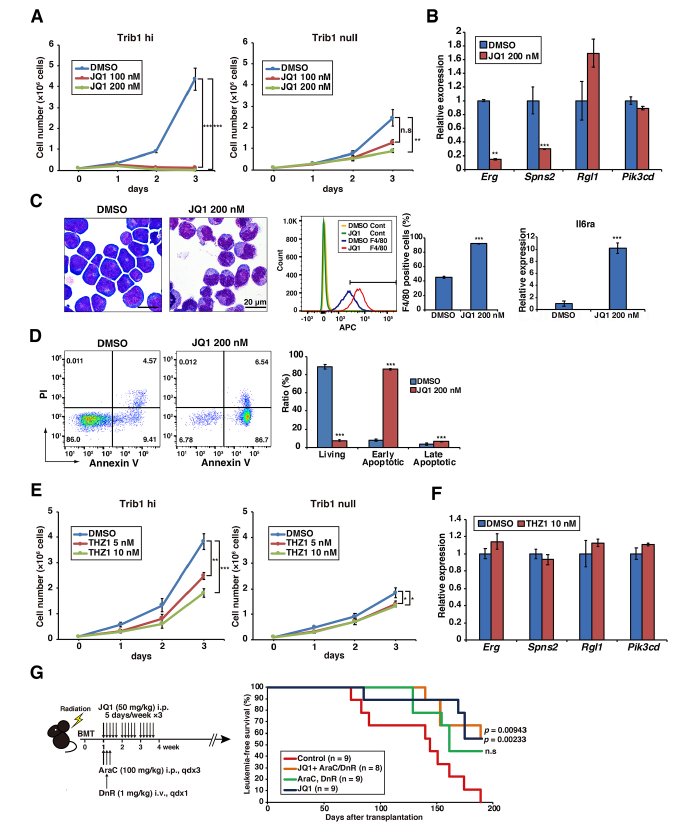
使用BRD4抑制劑JQ1處理Trib1 hi細胞, JQ1有效地以Trib1依賴的方式抑制白血病細胞的生長(A), 并抑制Trib1/Hoxa9靶向Erg和Spns2的表達,而不是Trib1 hi細胞中Rgl1和Pik3cd的表達(B),且JQ1能誘導Trib1 hi細胞的單核細胞分化和早期凋亡(C,D)。作者又檢測了CDK7/8抑制劑THZ1作為超級增強子靶向藥物的作用,與JQ1相比,THZ1只表現出溫和的抗增殖作用,而沒有觀察到Erg和Spns2的下調(E,F)。骨髓移植Trib1 hi細胞后,一周后給小鼠注射JQ1,無論是否使用柔紅霉素和阿糖胞苷治療,JQ1治療都能顯著提高白血病小鼠的存活率(G)。總的來說,這些結果突出了靶向Trib1修飾的Erg超級增強子的重要性。
7.TRIB1-ERG軸在AML細胞中的參與
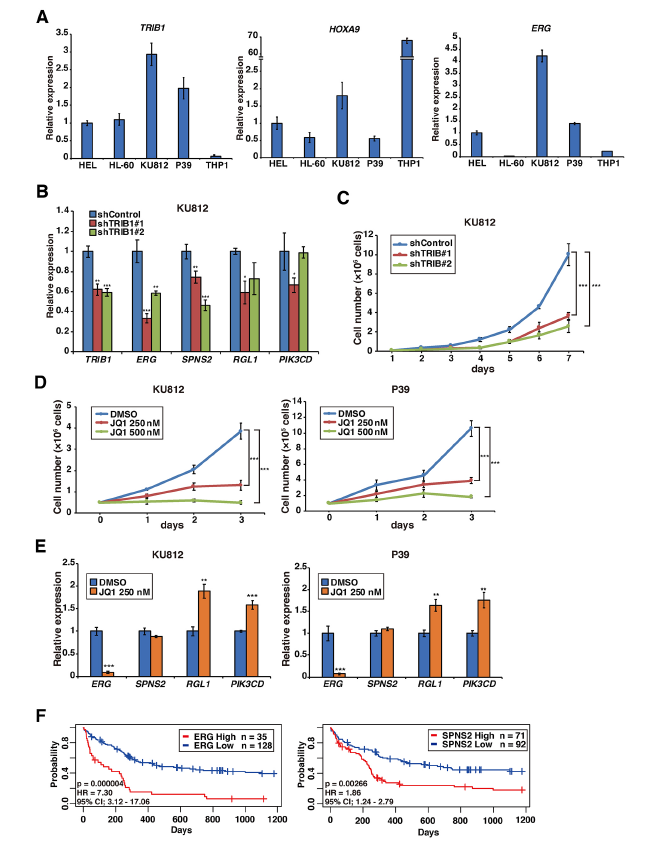
通過分析TRIB1和HOXA9在人AML細胞系中的表達,發現在KU812和P39細胞株中,TRIB1和HOXA9表達水平較高,HL-60細胞ERG低表達提示HOXA9對ERG表達的調控機制可能不同(A)。shRNA介導的TRIB1缺失發現KU812細胞中ERG和SPNS2的下調(B),KU812細胞的生長受到抑制(C)。利用250 nM的JQ1處理KU812和P39細胞,發現其對KU812和P39細胞均有生長抑制作用,并抑制了兩株細胞的ERG表達(D,E)。這些結果表明,Trib1介導的ERG上調在人類AML白血病細胞生長中發揮重要作用。通過預后掃描在線平臺重新評估了ERG和SPNS2上調在人類AML中的可能作用,發現ERG或SPNS2的表達水平與正常核型AML的不良預后顯著相關(F)。總的來說,我們的結果表明ERG是Hoxa9和C/EBP的一個重要靶點,并且可以被Trib1調控。
小結:
1.Trib1通過降解C/EBP和修飾與Hoxa9相關的超級增強子來影響Hoxa9在髓系白血病發生中的作用。
2. Erg是Trib1和Hoxa9的關鍵靶點,對BRD4抑制反應迅速,在白血病發生中起關鍵作用。
參考文獻:
Seiko Yoshino, Takashi Yokoyama, Yoshitaka Sunami, et al. Trib1 promotes acute myeloid leukemia progression by modulating the transcriptional programs of Hoxa9. Blood. 2020.